






安装GOplot
install.packages("GOplot")
激活GOplot
library(GOplot)
因为我喜欢用excel进行一些文本操作,所以也激活了操作xlsx格式的r包
library(openxlsx)
###########试运行帮助文档中的教程##########
help(package = "GOplot")
#点击Help Pages下的EC

这里有这个数据的一些描述,例如这是内皮细胞的转录组信息
详细的描述(有道翻译)
数据集包含来自两个稳态组织(大脑和心脏)的内皮细胞的转录组信息。更详细的信息可以在Nolan et al. 2013的论文中找到。将数据归一化并进行统计分析以确定差异表达基因。利用DAVID功能注释工具对一组差异表达基因(校正p-value<0.05)。
在来看一下EC数据里有哪些内容
data(EC)
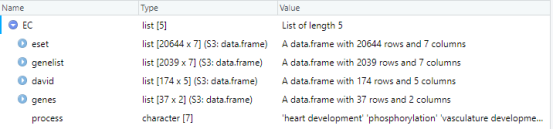
查看一下该数据集中各自的数据内容
head(EC$david)
也可以使用openxlsx的write.xlsx函数将这个参考数据包里面的信息下载下来。然后用excel查看。例如:
write.xlsx(EC$david,"EC-david.xlsx")
write.xlsx(EC$genelist,"EC-genelist.xlsx")

可以看到,david这个数据中包含174行*5列的内容,这5列分别是Category(GO富集分析中的三大生物学过程:BP、CC、MF),ID(富集到的GO号),Term(GO号对应的生物学过程名),Genes(该富集到该过程的genes),adj_pval(校正P值)。
查看一下genelist的内容
head(EC$genelist)
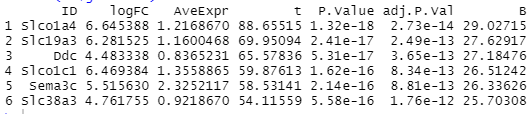
该数据一共2039行*7列,分别是ID(基因名),logFC(差异表达倍数),AveExpr(平均表达量),t(暂不清楚),P.Value(表达P值),adj.P.Val(校正P值),B(暂不清楚)。不清楚这个数据对作图是否有影响。因为后面需要用到chord_dat函数将这两个数据进行整合,见下图。下图中zscore值不知到和上面的t值和B值是否有关系。然后查看了帮助文档中对于circle_dat函数的使用要求。

显示term只要terms的category、ID、term、adj_pval和genes,genes只要ID和logFC即可。我们自己的数据根据以上要求修改好即可,但是列名必须和上述要求一致。就像下面这样。
EC-david-adjust.xlsx

EC-genelist-adjust.xlsx
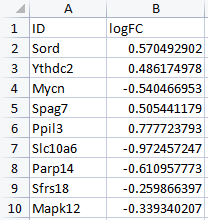
这里有一个问题,就是Genes列中基因名是大写的,而ID列中的基因名是小写的,这里在使用chord_dat函数时会报错,个人认为r会区分大小写。所以需要用Excel的UPPER函数将ID列的基因名转换为大写。这样就不会报错了。
读取准备好的这两个文件。
d1 = read.xlsx("EC-david-adjust.xlsx")
d2 = read.xlsx("EC-genelist-1adjust.xlsx")
#利用circle_dat()函数整合EC-david-adjust.xlsx和EC-genelist-adjust.xlsx。
circ = circle_dat(d1,d2)

可以看到,该函数将这两个数据进行了整合,即以每一个基因为目标分配各种信息。其中adj_pcal是由富集分析中的校正p值给出的。
d3 = c("heart development","vasculature development","blood vessel development","tissue morphogenesis","blood vessel morphogenesis")#这个是指定你要做那几个GO的弦图。这个必须于david文件中的名字一样。
然后使用chord_dat函数将circ、d2、d3进行整合。
chord = chord_dat(circ,d2,d3)
head(chord)

这个其实是生成了一个带有差异表达倍数的GO属性矩阵。
然后就可以开始出图啦。
pdf("chord3.pdf",height = 13,width = 13)#准备好一块画布。
GOChord(chord,space = 0.02,gene.order = 'logFC',gene.space = 0.25,gene.size = 5)#进行绘图。
dev.off()#然后必须关闭并保存这块画布。

然后就是可以进行一些微调啦,这张图片里用的基因有点偏多,可以在删除一些不太感兴趣的基因或者差异表达倍数不大或者padj值比较偏大的基因。















 1798
1798











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









