










Third Generation Sequencing – An Overview
More than 10% of current variants cannot be recovered until the human genome hit a contig N50 of 100kps. Current genes and gene blocks were not intact until the human genome hit a contig N50 of 1Mb. How do we get contigs large enough for good assemblies without spending a couple billion dollars?
The newest sequencing technologies are generating a lot of interest for this exact reason – long reads, up to 100kbs, can now be directly, or indirectly, sequenced. So what is the difference between these technologies, which is best for what applications, and how do I go about a genome assembly project in the era of third generation sequencing?
Short introduction to the technologies
There are several, but I’ll cover some of the current major players – PacBio, Nanopore, Illumina Synthetic Long Reads, 10x, Bionano, and Hi-C. The last two are more mapping technologies, so I have separated them as such.
第三代测序——概述
在人类基因组达到100kps的contig N50之前,超过10%的现有变异无法恢复。
直到人类基因组到达1Mb的contig N50,当前的基因和基因块才完整。
我们如何在不花费几十亿美元的情况下获得足够大的组装能力?
正是因为这个原因,最新的测序技术引起了人们的极大兴趣——长达100kbs的长读序列现在可以直接或间接地进行测序。
那么,这些技术之间有什么不同呢?哪些应用最合适?在第三代测序的时代,我该如何进行基因组组装项目?
技术简介
有几个,但我将介绍一些目前的主要厂商——PacBio、Nanopore、Illumina合成长读、10x、Bionano和Hi-C。
后两种是更多的映射技术,所以我将它们分开。
Sequencing Technologies:
PacBio is one of the most established long read technologies, having been around since 2010. The technology uses single-molecule real-time (SMRT) sequencing, which uses hairpin adaptors on a double stranded DNA fragment to make single stranded circular DNA template. This template is then loaded onto the SMRTcell chip (they REALLY like this acronym), where sequencing-by-synthesis occurs and the different light pulses emitted during synthesis of each nucleotide are recorded in 0.5-4h movies. Since the template is circular, the polymerase can continue multiple passes over the strand, which can then be split at adaptor sequences. The more passes, the more accurate the base calls.
PacBio是最成熟的长读技术之一,大约在2010年就出现了。该技术采用单分子实时(SMRT)测序,利用双链DNA片段上的发夹适配器来制作单链环状DNA模板。然后将该模板加载到SMRTcell芯片(他们非常喜欢这个首字母缩写词)上,通过合成进行排序,每个核苷酸合成过程中发出的不同光脉冲记录在0.5-4h的电影中。由于模板是圆形的,聚合酶可以在链上继续多次传递,然后可以在适配器序列上进行分裂。传球次数越多,基础调用就越准确。
Oxford Nanopore works by feeding a strand of DNA through an electrically resistant protein nano pore membrane. The sequencer reads the disruption in the current passed through the membrane as each base passes through. Since the bases have different electronic characteristics, each nucleotide has a different signature of disruption, allowing for base calls. Machine types (SmidgION, MinION, and PromethION) all vary mainly by size of the sensory and number of pores.
Illumina TruSeq Synthetic Long Read technology was originally billed as Moleculo, this is Illumina’s bid in the long read game. These aren’t real long reads, as the name implies. Input DNA is sheared into 10kb fragments, which are then assigned a barcode. Each 10kb fragment is sheared further, into traditional sized Illumina read pieces but with the barcode of the fragment it came from. This allows for the reassembly of up to synthetic 10kb stretches after (deep) sequencing.
10X (Chromium) works similarly to Illumina’s Synthetic Long Reads in that it uses barcodes to group and assemble short reads from a single large template. A small amount of high quality DNA is separated into droplets, each with 10 molecules of 100kb DNA. Emulsion PCRresults in short fragments from the long read template, each barcoded by droplet. These fragments are sequenced with Illumina HiSeq, and post processing bins and assembles the fragments. These fragments are then aligned with similar fragments to form large artificial reads. In contrast to Illumina Synthetic Long Reads, the copies of a fragment are split across different barcodes, yielding low read depth per molecule (0.2X) while maintaining phasing for each molecule. With a large number of molecules (~150 per region), the result is phasing information with 30X coverage (0.2X coverage per molecule x 150 molecules). More info (well done videos).
Mapping Technologies:
Bionano (Irys) is a unique departure from other sequencing technologies in that 1) it harnesses original gel capillary methods from the Sanger days and aims for low resolution - about 1kb; and 2) it’s really a mapping technology. Large fragments of DNA nicked at known sequence motifs (think restriction enzymes) and fluorescently labeled and annealed back together. The long, intermittently labeled fragments are fed into a chip, where the sequences flow through gel micro-grooves and (admittedly beautiful) photos are taken of the fragments as they pass through the chip. Post processing of the images assembles the fragments based on nick pattern and constructs long fragments with a pattern of the known nick sequence. This low resolution physical map of the DNA provides a useful scaffold for orientation, structural changes, and mapping of other read technologies.
The other major mapping protocol is Hi-C or the derived cHiCago, which leverages spacial information in chromatin to produce map information. Hi-C takes intact cells and cross links chromatin segments that are physically near each other. The chromatin is then cut with restriction enzyems and the ends of crosslinks sections marked with biotin and are ligated and the DNA is sheared. Biotin marked fragments (where ligation occurred) are pulled down and sequenced using Illumina. Based on the assumption that most proximate chromatin occurs in proximal portions of the same strand, sequence crosslink likelihood is a function of physical distance. More common co-occurance of sequence translates to likely more proximate (a twist on more likely recombined translating to likely more distant in traditional map creation). The output is large spans, sometimes entire chromosomal arms, which can detect orientation changes, inversions, etc. This technology is also very amenable to acting as a scaffold for other sequencing technologies. cHiCago is very similar, only that it artificially recreates the cross linking and eliminates the requirement for intact cells.
Comparison table for the technologies
See enormous attached table.
Important Considerations
While the above data is useful in comparing the different technologies, it is important to point out some considerations that are unique to the third generation technologies:
Read length:
Read length is no longer a generally tight range that we have grown to know and love in NGS. Read lengths (or read spans in mapping) occur along a distribution unique to each technology (i.e. PacBio has a long tail on the long read end). This ends up being highly critical - you need a diversity of read lengths to assemble a genome, especially on the longer end. Length is key here - remember, the glory of long read data is that we can now fill in gaps that result from repeats. Repeat distribution exponentially decreases as length increases in many eukaryote genomes – i.e. there are 300 more repeats that are 100bp long (difficult for Illumina to resolve) than there are repeats that are 3,650bp long (difficult for Tru-Seq to resolve). A focus on longer reads dramatically improves assemblies by bridging the majority of gaps and fixing misassemblies, orientations, etc (see Lee et al. Figure 6).
Take home: While lots of kbps of output from a PacBio run is awesome, focusing on the coverage in the 20kb range rather than total output may be more efficient. These are the reads that will really help clean up the assembly and give more complete data (see Lee et al. Figure 5)
Use what you have!
Illumina sequencing has been the largest market share of sequencing for years. All that data is still very useful - in synergy with the new long read technology. This is because Illumina sequencing is still the most accurate raw output of the current technologies and relatively cheap per bp - so much so Tru-Seq, 10x, and Hi-C all leverage it. Hybrid assemblers (Canu, MaSuRCa, etc.) allow for the combination of data input to reduce the cost of de novo assembly. Supplementing PacBio or Nanopore reads with Illumina short-reads reduces the long read depth needed to achieve reasonable accuracy while still spanning long gaps.
Why chose one technology?
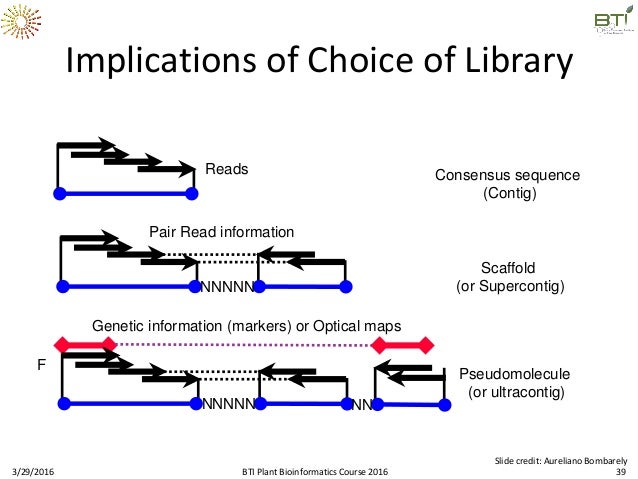
This generation of technology has seen popularity of both sequencing and mapping - which are very commonly used together. While long read sequencing provide long, bp scale resolution on fragments in the 10s of kbps… they don’t span chromosomal arms. There is no way to get orientation in relation to each other. Lower resolution mapping technologies like bionano or HiC provide longer range scaffolds and order of magnitude larger (100s of kbps). Adding short, accurate Illumina reads to an error prone, long PacBio scaffold results the best of both worlds - so does adding high resolution 10kb reads to a low resolution map of a chromosomal arm (illustrated here). Figure 1 of the Brickhart et al. 2016 Goat paper maps out how these technologies are used together.
Case study: Goat Genome
Researchers at the US Department of Agriculture and the National Human Genome Research Institute, used a combination of long-read sequencing, optical maps, scaffolding technology, and short-read sequencing to de novo assemble a goat reference genome that is 400 times more contiguous than the previously published assembly. It’s an interesting read - worth a look.
Table 1 from Brickhart et al. 2016. ARS1 is the PacBio + Optical Map + Hi-C + Bionano
"It's a night and day difference," Phillippy added. Contig size, contiguity, and overall accuracy are improved, he said.
Should you start caring about phasing?
Phasing of haplotypes in assemblies are becoming easier to obtain, especially with 10X and Illumina Synthetic Long Read (they really need a shorter name). With the increased availability of this information, it may be worth looking into the utility of such information to see if it would help in any of the analyses you have planned (it really is case by case).
10X has some awesome videos (see the two on linked reads) about the utility of this information, but the Illumina platform or possibly eventually Nanopore will offer the same output.
General Guidelines
Okay, so this is a lot of information and by all means not all of it. What are some general guidelines for approaching a de novo project? This paper (linked all over in this article) provides some really good insight, and this talk provides some really good comparisons (and a paper is upcoming). I will use their insights as I have personally little experience with these technologies as of yet.
1) “The highest quality genomes available have been assembled from the longest possible reads aided by the longest possible mapping… The per-nucleotide error rate of the reads have had little effect on the per-nucleotide assembled sequence accuracy, as well tuned algorithms can effectively reduce even 30% per-nucleotide error to below 1% with sufficient coverage.” – Lee et. al 2016
Translation: It is wise to split the budget to include both mapping and long reads. Coverage can mean more than raw accuracy, and you want at least some very long reads. Which technologies fit your project will be dependent on the project, but these should be your main goals.
2) “20X coverage of a genome should be enough to well assemble a genome, but we recommend researchers sample >75X when using the new long read sequencing technologies to make error correction steps more effective and to ensure high coverage is available for the longest reads… We recommend 20X coverage of error corrected reads over 20kbps long, using haploid or inbred samples if possible.” –Lee et al. 2016
Do note that the sequencing burden here can be dramatically reduced with hybrid assemblies, as Illumina reads can be used to correct for higher error rates in long reads. However, you still REALLY need REALLY long reads. Looking at some of the hybrid assembly papers as they come out may provide some guide as to how exactly to split between Illumina based, long read and mapping. Performing some simulations is also a really great idea, as they seem to match reality reasonably well (See Lee et al. Figure 3).
3) “For the human genome the read lengths need to average over 150kbp before complete chromosomes should be possible. If the historical trends continue, this could be achieved in as little as 3 to 4 years.”
While it may be tempting to jump on board the long read genome wagon, some of us will have to wait a little bit longer for this to be ideal. Larger genomes and genomes with weird characteristics (odd GC content, polyploidy, repeats, etc.) will be better served in the near future, when the technology matures a bit and more testing has been done. Looks like I’ll be waiting another couple years for a salamander genome :(.
4) Keep up with the reports. This isn’t just in journals, but also in the tech releases from the platforms themselves. Things are changing fast, and sometimes you can get in on early trials, find out about soon to be release technology that would be more efficient. Twitter is a good way to keep up with this, as is just routinely checking the sites.
5) When budgeting…keep in mind that informatics is now a large part of the time/money for a genome. These technologies are new and working through the kinks will take longer than you expect. If you don’t have the cash/expertise for this side of things, consider technologies that are designed for ease of use and analysis or waiting until things become a bit more established as far as best practices. Waiting a year to do an established workflow may save you time and money over going forward and paying someone to troubleshoot for a year.
Other analyses:
What about RNAseq? Microbiomes? Evolutionary Biology? Clinical research?
Dig in… it’s all changing! I don’t have specific expertise in any of this, but I’m always happy to help you wade through it all!
Sources
Sources for this article were mainly the links below, the vendors’ websites, and a lot of googling.
Further reading
Lee et al. 2016: http://www.biorxiv.org/content/biorxiv/early/2016/04/13/048603.full.pdf
Eisenstein 2015: http://www.nature.com/nbt/journal/v33/n5/fig_tab/nbt0515-433_T1.html
Goat Paper Summary: https://www.genomeweb.com/sequencing/goat-genome-demonstrates-benefits-combining-technologies-de-novo-assembly
Goat Paper Preprint: http://biorxiv.org/content/biorxiv/early/2016/07/18/064352.full.pdf
Koala combo assembly talk from PAG 2017: http://stream.dcasf.com/webinar/de-novo-sequencing-of-the-koala-genome/
A REALLY good talk about combination of technologies in de novo genomes from PAG 2017:
An article about the same study: https://www.genomeweb.com/sequencing/hi-c-services-launch-g10k-team-presents-head-head-sequencing-assembly-comparison
Useful Illumina metrics affecting assembly: https://www.illumina.com/Documents/products/technotes/technote_denovo_assembly_ecoli.pdf
















 1075
1075

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言












{kind=link}