








- 折叠转录本
- 分析目的:基于基因组比对结果,将相似的多转录本折叠成单个转录本(去冗余)
- PacBio分析软件:
- TAMA:https://github.com/GenomeRIK/tama
- TAMA简介
- TAMA(T ranscriptome A nnotation by M odular A lgorithms 是一款设计用于处理 Iso Seq 数据和其他长 reads 转录本数据,该软件 2019 年 在预印本在线期刊( bioRxiv )发表 。
Illuminating the dark side of the human transcriptome with TAMA Iso Seq analysis:https://www.biorxiv.org/content/10.1101/780015v1
- 三个主要应用:
- 1 TAMA Collapse (折叠冗余转录本
- 2 TAMA Merge
- 3 TAMA GO
- TAMA(T ranscriptome A nnotation by M odular A lgorithms 是一款设计用于处理 Iso Seq 数据和其他长 reads 转录本数据,该软件 2019 年 在预印本在线期刊( bioRxiv )发表 。
- 下载:
- git clone https://github.com/GenomeRIK/tama
- 安装:直接使用,无需编译
- 依赖环境:Python2、Biopython
- 使用命令:
- python /path-to-tama/tama_collapse.py -b BAM -s flnc.fasta.sorted.bam -f hg38.fa -x no_cap -e longest_ends -p isoform > tama.collapse.log
- 参数说明


- 结果展示
- 文件
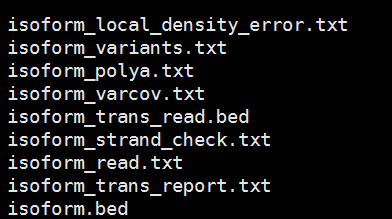
- 正链基因,右边相同,左边折叠;负链基因,左边相同,右边折叠


- 文件
- TAMA简介
- cDNA_Cupcake:https://github.com/Magdoll/cDNA_Cupcake
- 简介:
- cDNA_Cupcake是用于分析测序数据的 Python 和 R 脚本的集合,主要用于 cDNA
- 典型应用:
- 1 折叠转录本;
- 2 合并多样本结果
- 3 饱和度曲线
- 4 鉴定融合基因
- 下载:
- git clone https://github.com/Magdoll/cDNA_Cupcake.git
- 安装:
- cd cDNA_Cupcake
- git checkout origin/Py2_v8.7.x
- python setup.py build
- python setup.py install
- 使用命令:
- python /path to cDNA_Cupcake/cupcake/tofu/collapse_isoforms_by_sam.py
- --input flnc.fasta s flnc.fasta.sorted.sam
- --min coverage 0.99 min identity 0.95
- --max_5_diff 1000 max_3_diff 100
- -o prefix
- 参数说明:
- 结果展示:
- 结果文件
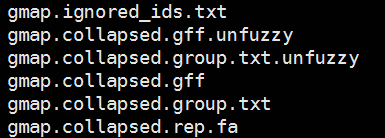
- 结果文件
- 简介:
- TAMA:https://github.com/GenomeRIK/tama
- 基因/转录本鉴定
- SQANTI3:https://github.com/ConesaLab/SQANTI3
Corrigendum: SQANTI: extensive characterization of long read transcript sequences for quality control in full length transcriptome identification and quantification:https://genome.cshlp.org/content/28/7/1096
- 简介:
- SQANTI3是SQANTI 工具(发布)的最新版本,该工具合并 SQANT 1 和 SQANTI2 中的功能并添加新的功能 ,用于全长转录本的深度表征 。
- 主要应用:
- 1 sqanti3_qc.py (执行转录本的深度表征
- 2 sqanti3_RulesFilter.py (基于机器学习算法过滤假阳性转录本
- 下载:
- git clone https://github.com/ConesaLab/SQANTI3.git
- 安装:
- 1 export PATH=$HOME/anacondaPy37/bin:$PATH
- 2 conda update conda
- 3 cd SQANTI3
- 4 conda env create f SQANTI3.conda_env.yml
- 使用命令:
- ## step1: qc
- export PATH=/local_data1/rna_training/liwei/software/anaconda3/bin:$PATH
- export PYTHONPATH=$PYTHONPATH:/local_data1/rna_training/liwei/software/cDNA_Cupcake-9.1.1/sequence/
- export PYTHONPATH=$PYTHONPATH:/local_data1/rna_training/liwei/software/cDNA_Cupcake-9.1.1/
- source activate SQANTI3.env
- python /local_data1/rna_training/liwei/software/SQANTI3/sqanti3_qc.py
- test_chr13_seqs.fasta Homo_sapiens.GRCh38.86.chr13.gtf Homo_sapiens.GRCh38.dna.chromosome.13.fa
- --fl_count chr13_FL.abundances.txt -o Sample
- 参数说明
- ## step2: filter
- export PATH=/local_data1/rna_training/liwei/software/anaconda3/bin:$PATH
- export PYTHONPATH=$PYTHONPATH:/local_data1/rna_training/liwei/software/cDNA_Cupcake-9.1.1/sequence/
- export PYTHONPATH=$PYTHONPATH:/local_data1/rna_training/liwei/software/cDNA_Cupcake-9.1.1/
- source activate SQANTI3.env
- python /local_data1/rna_training/liwei/software/SQANTI3/sqanti3_RulesFilter.py
- Sample_classification.txt Sample_corrected.faa Sample_corrected.gtf
- 参数说明

- ## step1: qc
- 结果展示:
- 简介:
- TAPIS:https://bitbucket.org/comp_bio/tapis/src/master/(没有更新,存在bug不推荐)
- SQANTI3:https://github.com/ConesaLab/SQANTI3
- 实操分析
- SQANTI3测试数据(example文件夹)
- 文件

- 文件
- 补充:SQANTI3 sqanti3_qc.py )分析的第一个输入文件来源于 collapse 之后的结果( gmap.collapsed.rep.fa
- SQANTI3测试数据(example文件夹)
全长转录组之基因和转录本鉴定
最新推荐文章于 2024-06-02 09:55:02 发布




















 3155
3155











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









