









Single-Molecule Sequencing Assists Genome Assembly Improvement and Structural Variation Inference
Open ArchivePublished:April 19, 2016DOI:https://doi.org/10.1016/j.molp.2016.04.002
Dear Editor,The single-molecule real-time (SMRT) sequencing platform presented by Pacific Biosciences (PacBio) is regarded as a third-generation sequencing technology (Eid et al., 2009, Roberts et al., 2013). PacBio delivers long reads from several to tens of kilobases (kbs), which are ideal for filling unsequenced gaps due to unusual sequence contexts, such as high-GC content or repeat-rich regions (Bashir et al., 2012, Berlin et al., 2015, Chaisson et al., 2015). PacBio long reads are also favorable for detecting large DNA fragments harboring structural variations (SVs), such as inversions, translocations, duplications, and large insertions/deletions (indels) (Ritz et al., 2010, English et al., 2014). However, one drawback of PacBio is the high error rate of base calling for single pass coverage of the genome (Au et al., 2012, Koren et al., 2012). This drawback can be mitigated by increasing sequencing coverage to achieve high consensus accuracy, but the requirements may be prohibitive for the de novo assembly of large- or medium-size genomes using only PacBio when considering both budgetary and computational costs. Alternatively, PacBio may be used for assembly improvement of near-finished reference genomes, especially for filling gaps in which unsequenced bases are represented by the letter N (English et al., 2012). Here, we combined PacBio (∼15x) with Illumina reads (∼40x) to improve the genome assemblies of African wild (Oryza barthii) and cultivated rice (O. glaberrima), and to infer large SVs between O. barthii and O. glaberrima.
The two African rice genomes were first sequenced using Illumina to produce ∼153 million and ∼120 million 100-nt paired-end short reads for O. barthii and O. glaberrima, respectively. The PacBio RS-II system was used later to generate long reads. On average, each SMRT cell generated ∼80 000 usable long reads (read quality ≥0.85; subread length ≥100 bp) with an average length of 5.8 kb and a total base yield of 455 Mb, covering the rice genome at least one-fold per SMRT cell. To fully use every segment of the PacBio reads, the assembly improvement process was conducted over five steps (Supplemental Figure 1 and Supplemental Notes). Using the released O. barthii and O. glaberrima genomes as references, the pipeline generated two new assemblies that appeared to be greatly improved (Supplemental Table 1). At the contig level, the original 25 409 O. barthii contigs were reduced to 3448; the total length increased from 305.7 Mb to 325.0 Mb, and N50 increased from 18.9 kb to 201.3 kb. As for O. glaberrima, the 25 481 contigs were reduced to 5844; the total length increased from 303.3 Mb to 316.5 Mb, and N50 increased from 23.2 kb to 195.1 kb. At the chromosome level without counting the Ns, the total sequence of O. barthii increased from 305.7 Mb to 324.5 Mb, and that of O. glaberrima increased from 273.6 Mb to 294.5 Mb. In addition, the gaps in the full chromosome assemblies of the two genomes were effectively filled: in O. barthii, 12 065 gaps were filled, reducing Ns from 2 539 700 to 32 796 bases; in O. glaberrima, 14 847 gaps were filled, reducing Ns from 13 084 265 to 29 389 bases.
To assess the assembly coverage, we aligned Illumina and PacBio reads to the original and improved genomes. Percentages of unique and multi-mapped Illumina reads and average sequencing depths were all slightly increased in both genomes (Supplemental Figure 2A). The consensus map generated by Illumina alignments showed slightly decreased depths at 1×, 4×, 10×, and 20× coverage, suggesting that a part of the filled gaps or newly scaffolded contigs was attributed to the PacBio reads (Supplemental Table 2). We also noticed 4.4 (2.0%) and 3.5 (2.4%) million more properly paired mapped reads in the improved O. barthii and O. glaberrima assemblies, respectively, compared with the alignment in the original assemblies. Assembly improvement was also reflected in PacBio alignment by BLASR (Chaisson and Tesler, 2012) (Supplemental Table 3 and Supplemental Figure 2B). Mapping rates of the ∼2.5 million PacBio reads in the original and improved genomes increased from 90.0% to 92.1% for O. barthii and from 92.5% to 93.0% for O. glaberrima, respectively. The consensus map built from PacBio read alignment showed that 89.8% and 97.0% of the improved assemblies were covered by at least one PacBio read in O. barthii and O. glaberrima, respectively. However, approximately 610 Mb and 535 Mb respective unmapped reads in O. barthii and O. glaberrima were not assembled into the genomes.
We then used RepeatMasker to identify repetitive sequences on both original and improved assemblies to compare repeat contents. Approximately 95.8 Mb (88.6 Mb in the original) of the 325.5 Mb (29.4%) improved O. barthii genome was identified as repeats; in the O. glaberrima genome, 103.2 Mb (96.7 Mb in the original) of the 318.6 Mb (32.4%) improved genome was classified as repeat (Supplemental Figure 2C). Inspection of the gap-filled regions revealed that, whereas repeat percentages increased 10.35% and 19.02% in O. barthii and O. glaberrima, respectively, the GC content in the filled gaps of O. glaberrima did not substantially change. When we examined the GC content of the reads that filled the gaps, we found that the majority in O. barthii had extraordinarily high GC content, peaking at 70-80%; in O. glaberrima, the major peak of GC content fell between 20% and 40% (Supplemental Figure 3). Therefore, unsequenced gaps in the two genomes are likely due to different reasons, but PacBio reads are seemingly robust in covering GC-rich, AT-rich, and highly repetitive sequences.
We used PacBio long reads to detect large SVs between the two rice genomes (Ritz et al., 2010, English et al., 2014). SV inference was performed by PBhoney (English et al., 2014) to initially identify a rough location of a putative SV event, followed by our custom pipeline to validate and determine the exact locations of an SV event (Supplemental Notes). PBhoney includes a “spot mode” to detect small SVs in lengths of dozens to hundreds of bases located within in one single long read and a “tail mode” to detect large indels, inversions, and translocations inferred from a set of reads were distantly mapped to different genomic locations. By the spot mode after removing background noise, the pipeline detected a total of 1894 insertions (DNA loss in O. barthii) and 2058 deletions (DNA loss in O. glaberrima) based on the alignment of O. glaberrima reads against the O. barthii genome; conversely, 302 insertions (DNA loss in O. glaberrima) and 2064 deletions (DNA loss in O. barthii) were detected based on the alignment of O. barthii reads against the O. glaberrima genome. However, because both assembled genome sizes were still smaller than their estimated sizes (325 Mb assembled versus 411 Mb estimated for O. barthii; 294 Mb assembled versus 354 estimated for O. glaberrima) (Jacquemin et al., 2013), we were concerned that a large proportion of the indels might have been due to incompleteness of the current assemblies. Thus, we applied an additional criterion to increase filtering stringency so that an indel event must have mutually occurred in both genomes, namely, a detected insertion in O. barthii must correspond to a deletion in O. glaberrima at the same genomic locations to guarantee a bona fide SV. This filtering resulted in a total of 181 indels between the two genomes, including 79 deletions in O. glaberrima and 102 deletions in O. barthii. However, the majority of these indel events occurred in either intergenic or intronic regions, with very few occurring in coding sequences. The spot mode usually detects indel events of hundreds of bases occurring in the same reads and is thus relatively accurate. The same pipeline run on the original assemblies detected 52 of 79 deletions in O. glaberrima and 51 of 102 deletions in O. barthii, respectively (Figure 1A ). By the tail mode, 110 deletions, six insertions, 156 inversions, and 51 translocations based on the alignment of O. barthii reads to the O. glaberrima genome were found, and 313 deletions, 27 insertions, 248 inversions, and 336 translocations based on the alignment of O. glaberrima reads to O. barthii genome were found (Figure 1B). Considering that O. barthii is the progenitor genome of O. glaberrima, it is reasonable that nearly three times more deletion events were detected in O. glaberrima than in O. barthii. In addition, the average length for spot-mode deletion is 234 bp, whereas for the tail-mode deletion, the average length was 7.98 kb, with the longest at 180 kb, reflecting the advantage of the PacBio platform in detecting large SVs (Figure 1C). As exemplified in Figure 1D, an exon of a gene encoding the PPR repeat domain appears to be entirely missing due to a ∼600-bp deletion from O. glaberrima, according to the alignment of O. glaberrima reads against the O. barthii genome (Figure 1D).
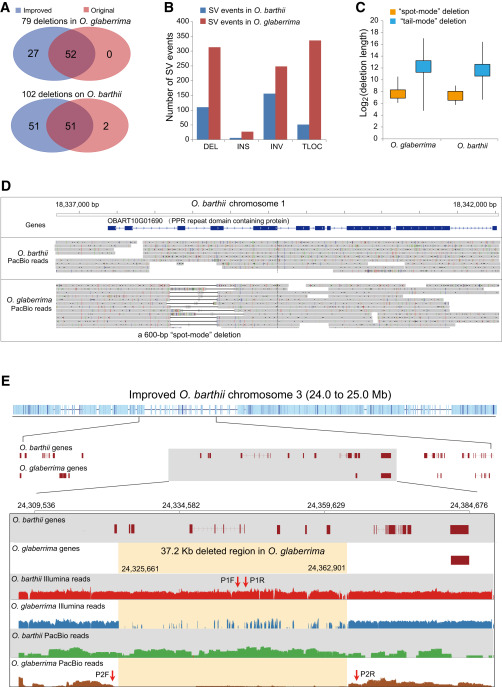
Figure 1Structural Variations Detected between O. barthii and O. glaberrima Based on PacBio Long Reads.
The combinatorial resequencing data of the two African rice genomes gave us the chance to examine large SVs between the two genomes. From the list of gene-associated SVs in our data, a 37.2-kb region between 24 325 661 bp and 24 362 901 bp on O. barthii chromosome 3 was completely deleted from O. glaberrima, covering six protein-coding genes (Figure 1E). To experimentally validate this large deletion in O. glaberrima, we designed two pairs of primers; while one pair was designed within the candidate fragment in the O. barthii genome, the other pair was designed at the edges of the deleted fragments in the O. glaberrima genome (Figure 1E). The P1 primers designed within the 37.2-kb fragment successfully amplified the DNA from O. barthii but failed in O. glaberrima, whereas the P2 primers designed at the right and left edges of the deletion successfully amplified the DNA in O. glaberrima but failed in O. barthii (Supplemental Figure 4). Both ways proved that the 37.2-kb deletion indeed existed in O. glaberrima. Nevertheless, whether this SV is associated with domestication requires further investigation. In terms of applying PacBio in crop improvement and molecular breeding, ab initio sequencing a large crop genome is seemingly unfeasible at the current stage, but this technology provides a practical solution for initial survey of crop germplasm or genetic mapping of qualitative trait loci involving large SVs that prohibit designing a molecular marker for fine mapping of candidate genes. Our study illustrates the potential of PacBio single-molecule sequencing in expediting the process of gene discovery for crop improvement and downstream functional validation of candidate genes.
Funding
This work was supported by the National Basic Research Program of China (973 Program) ( 2012CB910900 ), and the National Natural Science Foundation of China ( 31330048 and 31471457 ).
Author Contributions
W.X. and Z.Z. conceived the project; Z.Z., M.H., and L.S. performed experimental validation; M.Q. performed PacBio sequencing; Y.J. and W.X. performed assembly improvement, structural variation analyses, and other bioinformatics analyses; G.Z. and H.H. performed SNP analyses; W.X. and Z.Z. wrote the manuscript.
Acknowledgments
No conflict of interest declared.
















 308
308











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言












{kind=link}
{kind=link}