










Hybrid error correction and de novo assembly of single-molecule sequencing reads
混合误差校正和重新组装的单分子测序读取
单分子测序仪可以产生多千位酶序列,具有极大的改善基因组和转录组组装的潜力。然而,单分子测序的错误率很高,这限制了它们迄今为止在细菌测序中的应用。为了解决这一局限性,我们引入了一种修正算法和装配策略,该算法使用短的、高保真度的序列来纠正单分子序列中的错误。我们演示了这种方法在PacBio RS仪器从噬菌体、原核和真核全基因组(包括鹦鹉Melopsittacus undulatus之前未测序的基因组)产生的读谱(reads)以及玉米(Zea mays)转录组的RNA-Seq读谱(RNA-Seq reads)上的实用性。我们的长读校正达到了>99.9%的基调用精度,导致了比当前测序策略更好的程序集:在最好的例子中,相对于高覆盖率的第二代程序集,中位数叠置的大小增加了五倍。如果读长度继续增加,包括单叠叠基因组细菌染色体组装的前景,预计会有更大的收益。
第二代测序技术,从2004年的454个焦磷酸测序开始,到2007年的Illumina合成测序2和其他技术,通过降低成本和成倍增加的产量,已经彻底改变了DNA测序。尽管第二代仪器提供的收益很大,他们有几个缺点。首先,它们需要在测序前对源DNA进行扩增,从而导致扩增人工技术和与DNA5的化学-物理特性相关的基因组的偏倚覆盖。第二,当前技术生产相对较短的读取,中等长度的Illumina公司100个基点(max 150bp),454(700bp)最大可达1000bp)。短读使装配和相关分析变得困难,理论模型表明,将读长从1000bp降低到100 bp可能导致连续性下降6倍或更多
太平洋生物科学公司最近发布了他们的第一个商用“第三代”测序仪PacBio RS:一个实时单分子测序仪。它的目的是解决上述问题,不需要扩增和减少成分偏差,产生长序列(例如,median= 2,246,max= 23,000bp)使用最新的PacBio化学)和支持一个短的周转时间(24小时,样品测序)。长阅读长度将有利于从头开始基因组和转录组组装,因为它们有潜力解决复杂的重复和跨越整个基因转录本。然而,仪器产生的读数平均只有82.1%(ref.8)-84.6%(ref.9)的核苷酸准确度,均匀分布的错误主要由点插入和删除(Supplementary Fig 1)。这种高错误率模糊了读序列之间的比对,并使分析变得复杂,因为两个读序列之间的成对差异大约是它们各自错误率的两倍,而且远远超过大多数基因组汇编器所能容忍的5-10%的错误率;仅仅增加传统汇编器的对齐灵敏度在计算上是不可行的(Supplementary Fig Table1和Supplementary Fig.2和3)。此外,PacBio技术使用发夹适配器对双链DNA进行测序,如果测序反应同时处理两条DNA链(首先是正向,然后是反向),则可能导致嵌合读取。虽然可以生成准确的序列PacBio RS多次通过阅读一个环状分子(圆形共识或CCS),这种方法可以减少阅读倍长度等于分子是遍历的次数,导致更短的读取(例如,median= 423bp,max= 1915bp)。因此,如果错误率可以通过算法来管理,那么长时间的单遍读取具有很大的潜在优势。
为了克服单分子测序数据的局限性,释放其重新组装的全部潜力,我们开发了一种方法,利用短的、高精度的序列来纠正长单分子序列固有的错误(Fig 1)。我们的PBcR (PacBio纠正读)算法是Celera Assembly程序的一部分,它首先将短读序列映射到单个长读序列,并计算一个高度精确的混合一致序列,从而对单个长读序列进行修剪和纠正:将读取精度从低至80%提高到99.9%以上。修正后的混合PBcR读取可以单独重新组装,与其他数据组合,或导出用于其他应用程序。如下面的几个重要基因组所示,包括鹦鹉Melopsittacus undulatus之前未测序的1.2 gbp基因组,与第一代或第二代测序相比,使用这种方法整合PacBio数据可以大大提高装配质量,预示着“第三代”测序和组装的前景。
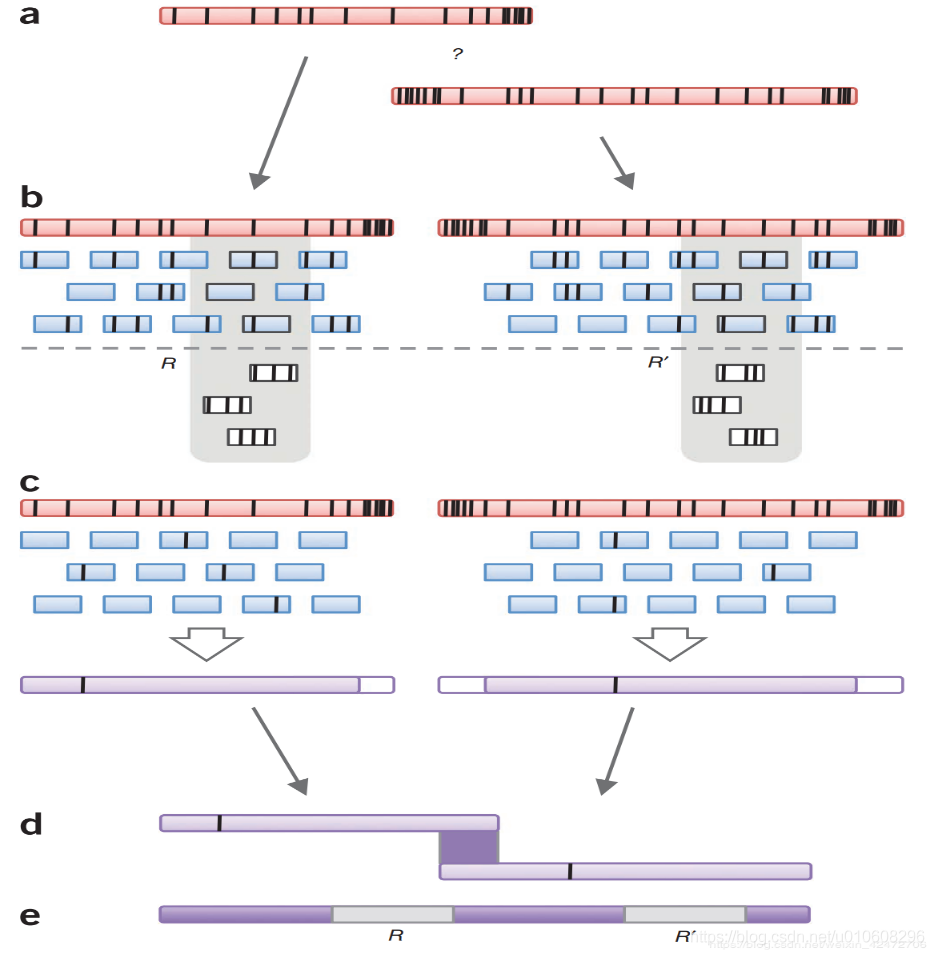
Figure 1 PBcR单分子读校正与装配方法。(a)在单遍PacBio RS读取(粉红色矩形)中,黑色竖条表示的错误使很难确定读取是否重叠。(b)将高保真度短读调整为容易出错的长读。准确的比对是可以计算的,因为一个短的、高精度的序列(约99%与事实相同)和一个PacBio RS序列之间的误差是两个PacBio RS序列之间误差的一半。在本例中,短读中的黑色条表示映射错误,这是长读和短读中的排序错误的组合。此外,存在两份不精确的重复(用灰色表示),导致每次复制时的读取量堆积。为了避免将读取映射到错误的重复复制,该算法选择一个截止值C,并且对于每个短读取只使用顶部的C次命中。伪映射(白色)被丢弃。
(c)其余的对齐用于生成新的一致序列(紫色),每当短读平铺中出现缺口时,就对长读进行修剪和分割。测序错误,黑色表示,可能传播到PBcR读取罕见的情况下,测序错误同时发生。(d)校正后,较长的PBcR序列之间容易出现重叠。(e)产生的组装集能够跨越仅使用短读就无法解决的重复。
结果
长读的重新组装
基因组组装是通过测序读取来重建基因组的计算问题。它与从头转录组组装密切相关的问题是基因组学的关键工具,需要从无数的短片段中进行排序。装配的问题通常是制定的找遍历图来源于测序读使用overlap-layout-consensus(OLC或String Graph)范例,图是由重叠的序列读取,或者De Bruijn图公式,图形是由给定长度的子字符串k来源于阅读。装配图的复杂度由排序误差和重复量决定,但重复量是所有装配算法和排序技术的最大障碍。在de Bruijn图公式下,重复时间超过k个碱基对形成分支节点,这些分支节点必须通过线程读取图或应用其他约束(如配对关系)来解决。相反,只有重复长度大于l=r- 2×o(其中r为读取长度,o为最小可接受重叠长度)时,才会在字符串图中产生未解析的分支。对于短读序列,k和l非常相似,因此对应的图几乎是等价的。但是,对于长读操作,l可能远远大于k的可行值。因此,长序列在简化OLC装配问题方面具有很大的潜力。在最极端的情况下,如果所有的重复都被更长的读取所跨越,那么基因组的OLC组装成其组成染色体和/或质粒将是微不足道的。在实践中,较长的读操作增加了跨越重复和检测重叠的概率,从而在较低的测序覆盖率下生成比较短的读操作更好的程序集。
作为一个简单的测试,我们使用高精度短读测序技术,在纠正lambda噬菌体PacBio RS序列中的错误后,评估了多个装配器的性能(Supplementary Table1);只有OLC组装器产生了一个叠叠序列。为了测试增加读长所带来的好处,我们模拟了酿酒酵母S228c基因组中不同长度的无错误数据,并比较得到的N50叠叠基因组大小(N,其中50%的基因组包含在≥N的叠叠基因组中,Fig2)。OLC程序集对于较长的读取变得越来越强大,显示随着读取长度的增长,重叠组大小几乎呈线性增长。相比之下,de Bruijn程序集由于图结构的固有限制和读线程问题的复杂性,在不增加k超出实际值的情况下,无法有效地利用长读。因此,我们开发了一种利用OLC方法对PacBio RS序列进行校正和装配的算法。
校正精度和性能
我们评估了PBcR校正和装配算法对Illumina、454和PacBio测序仪生成的多个短、长读数据集的校正和装配算法,包括三个具有可用参考序列的数据集:Lambda NEB3011, Escherichia coli K12, S. cerevisiae S228c(Supplementary Table2).经过50个高同一性序列后,校正精度和装配连续性均呈递减趋势;我们建议在性能和准确性之间进行折衷(Supplementary Table4 and 5)。使用Illumina的50个数据对每个参考生物的PacBio读进行校正,长读的准确性从~85%提高到>99.9%,嵌合和不正确修剪的reads分别测量到<2.5%和<1%(Table 1和(Supplementary Fig6)。这对于删除适配器序列是必要的,否则很难识别(在线方法)。
因此,校正后的读数比原读数略短,但长度没有受到显著影响(例如,校正前的中位数为848,校正后的中位数为767)。在纠正期间,由于异常低的质量或较短的长度,读操作也可能被丢弃,而成功纠正的读操作的百分比称为通量。所观察到的通量通常在60%左右,但是根据单个运行的质量差异很大。例如,S. cerevisiae S228c的读取量显得异常低,这可能是因为大部分测序是在冷泉港实验室测试期间使用预释放PacBio RS仪器完成的。然而,在所有情况下,校正算法都成功地识别出可用的数据,并输出高精度的长读。
混合从头组装
我们评估了PBcR读序列对全基因组组装的影响,无论是单独的还是与互补读序列结合。除了Celera汇编器之外,还有两个汇编器支持PacBio读取:ALLPATHS-LG和ALLORA。然而,两个程序都不执行纠正或重新组装从未纠正的读取。相反,ALLPATHS-LG使用原始读取来辅助脚手架和短读de Bruijn程序集的间隙关闭。这种方法的缺点是,短读的contigs中引入的错误可能得不到纠正,并且由于计算的限制,该函数仅适用于Illumina付费端文库<200 bp和Illumina远程跳转文库都小于10 Mbp的基因组。只有这里展示的鹦鹉基因组包含Illumina和PacBio reads的必要组合,但它的大小超过了限制,无法进行评估。基于AMOS20 22的长读汇编程序ALLORA,在计算上仅限于小基因组,并且需要高精度的PacBio序列(如CCS)才能运行。受我们最初研究结果的启发,其他研究人员使用我们的consensus模块手工纠正了2011年德国大肠杆菌爆发时的低准确度PacBio序列,并使用ALLORA9对其进行了迭代组装。我们现在已经评估了我们对同一大肠杆菌C227-11基因组的自动校正和装配方法,并发现它优于以前发表的装配(Table2)。
结论
目前的重新组装者无法有效利用现有单分子测序技术产生的长读测序数据,主要原因是误码率较大。我们的方法利用这一技术,以更短的、高身份序列作为补充,从而得到长、准确的转录本和改进的组装。由于我们的方法产生的平均叠架大小与读取长度相关,预计随着该技术的读取长度的提高,装配结果将会得到改善。该策略还得益于多种技术的互补性,当Sanger测序与第二代数据相结合时,后者首次可用,这证明了其强大的功能。我们的混合方法的结果是更高质量的装配与更少的错误和缺口,这将降低基因组整理的昂贵成本,并使更准确的下游分析。
高质量的装配体对基因组学的各个方面都至关重要,特别是基因组注释和比较基因组学。例如,许多微生物基因组分析依赖于已完成的基因组,但由于完成的成本与原始装配中的间隙数量成正比,因此产生已完成的序列仍然是令人望而却步的。真核基因组学需要连续的装配来捕获长、多外显子基因,并确定基因组组织和结构多态性。此外,最近的研究表明,在发现大的结构变异时,重新组装可能优于读图方法,即使有参考基因组可用。这对于理解癌症基因组的遗传变异和其他人类疾病,如经常包含基因融合、拷贝数变异和其他大规模结构变异的自闭症,具有特别重要的意义。很明显,高质量的程序集,具有长时间连续的叠架,将对广泛的学科产生积极的影响。
PBcR算法的潜在改进包括添加一个gap-closure例程来填充使用PacBio读取的短读数据中的排序空白,以及在一致调用期间合并单分子碱基调用。这对于第二代测序仪往往不能充分表达的GC-rich的序列,以及具有严重覆盖率波动的元基因组和扩增样本尤其重要。非均匀覆盖还需要对重复分离算法进行修改,因为目前的启发式假设一致的长读覆盖和错误。这可以包括更好地利用配对端信息或变异聚类,这也可以应用于单倍型分离问题。
我们已经证明,高错误率不一定是组装的障碍。高错误率、长读序列与互补的短读序列可以有效地组合在一起,产生任何现有技术都无法实现的装配,这使我们离“一条染色体,一条重叠群”的目标又近了一步。利用PacBio和其他技术,如离子激流技术,快速的周转时间将使生产高质量的基因组装配成为可能,只需所需时间的一小部分。未来的研究需要探索现有技术的相对成本和权衡,但从我们的研究结果中,我们预计未来的测序项目将包括长读和短读测序的组合。如今,短读插入库≥9 Kbp对于有效的远程搭建是必要的,而当前的PacBio读取只能提供有限的帮助。然而,如果单分子技术继续发展,以合理的成本和产量,读序列开始超过典型的细菌重复序列(~6 Kbp)的长度,一些细菌染色体的单叠基因组组装将成为可能,而不需要昂贵的对文库。此外,我们相信许多长期寻求的功能将被启用,如真核生物的单倍型分离、准确的转录组注释和真正的比较基因组学,这些功能将超越以外显子为中心的观点,包括整个基因组。















 755
755











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











