







前面介绍过fasq,fastq,bam三种生物数据分析中常用的格式。fasta一般作为比对的参考序列,fastq为测序数据,将fastq比对到fasta则生成bam文件,对bam进行排序建立索引,就可以直接输出为vcf格式,这个系列我们来详细介绍一下vcf格式的操作。
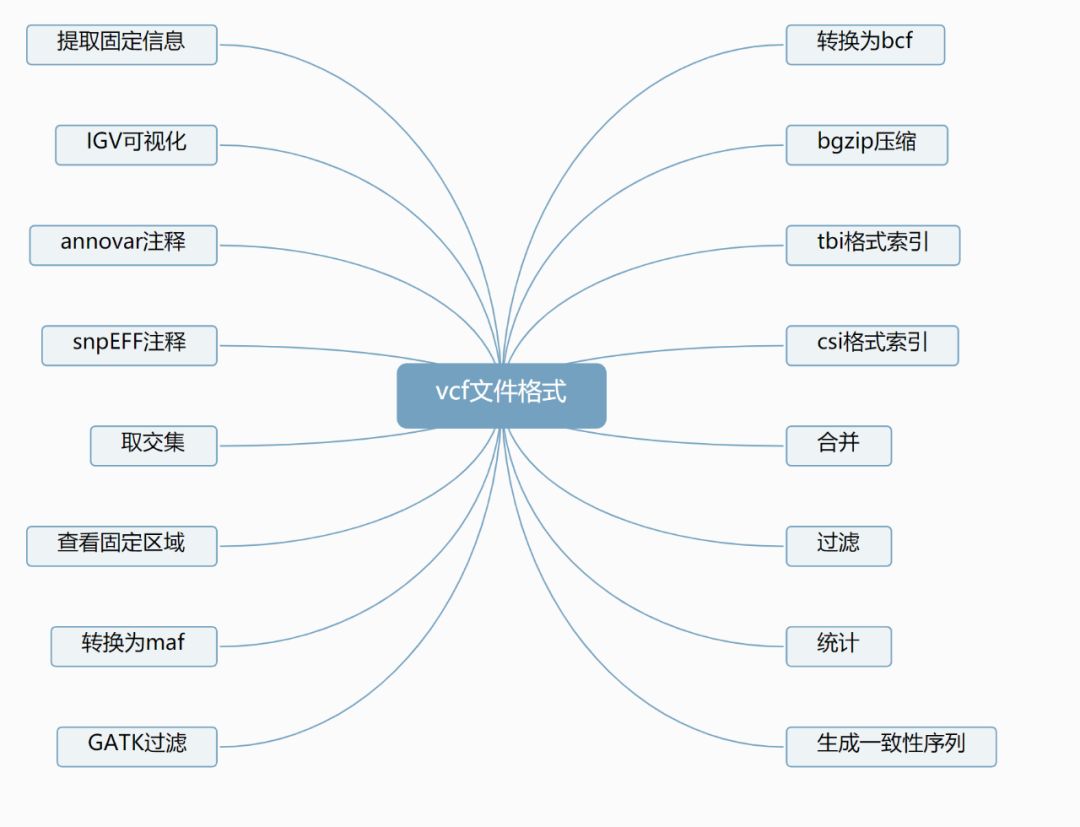
1 文件格式介绍
VCF是Variant Call Format的简称,是一种定义的专门用于存储基因序列突变信息的文本格式。在生物信息分析中会大量用到VCF格式。例如基因组中的单碱基突变,SNP, 插入/缺失INDEL, 拷贝数变异CNV,和结构变异SV等,都是利用VCF格式来存储的。将其存储为二进制格式就是BCF。
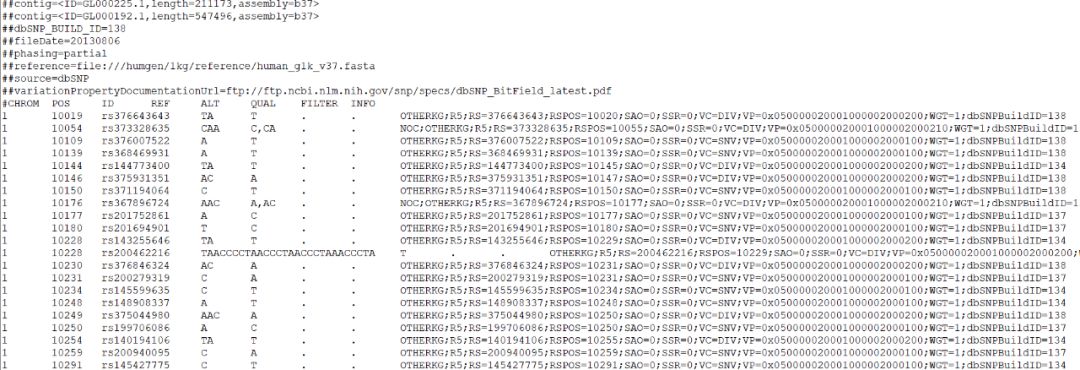
1.CHROM [chromosome]:染色体名称,
2.POS [position]:参考基因组突变碱基位置,如果是INDEL,位置是INDEL的第一个碱基位置。
3.ID [identifier]:突变的名称,
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









