







Align
align首先执行序列比对,然后进行结构叠加,进行多次迭代以便进行微调,在蛋白序列相似性大于30%的时候可以达到良好的效果。
用途
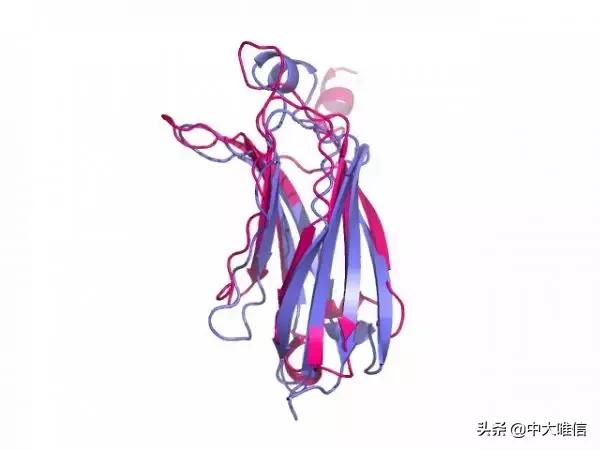
Align常常在结构生物学以及虚拟筛选中使用,当对不同的蛋白结构并对其进行比较时,我们就可以使用align比较蛋白结构,查看两者之间的差异,这个结构上的差异有一个量化的指标就是RMSD。它的概念和计算方式,都会在下面列出。目前,pymo是一个很流行的三维蛋白结构显示工具。本次的目的是,使用pymol对蛋白结构进行align,结果可以通过肉眼观测或者RMSD进行量化。
用法
align mobile, target [, cutoff [, cycles [, gap [, extend [, max_gap [, object [, matrix [, mobile_state [, target_state [, quiet [, max_skip [, transform [, reset ]]]]]]]]]]]]]
解释:
mobile =字符串:需要移动的对象名 target =字符串:目标的对象名 cutoff = 浮点数:截断值,默认2.0 cycles =整数:最大循环数,默认5 gap, extend,
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 2230
2230











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









