






hh-suite使用教程(翻译: 原文)
HHsuite 工具简要教程
- hhblits:迭代地使用查询序列或 MSA 搜索 HHsuite 数据库
- hhsearch:使用查询 MSA 或 HMM 搜索 HHsuite 数据库
- hhmake:从输入 MSA 构建 HMM
- hhfilter:按最大序列标识、覆盖率和其他标准过滤 MSA
- hhalign:计算两个 HMM/MSA 的成对对齐等
- hhconsensus:计算 A3M/FASTA 输入文件的一致序列
- reformat.pl:重新格式化一个或多个 MSA
- adds.pl:将 PSIPRED 预测的二级结构添加到 MSA 或 HHM 文件
- hhmakemodel.pl:从 HHsearch 或 HHblits 结果生成 MSA 或粗略 3D 模型
- hhmakemodel.py:从 HHsearch 或 HHblits 结果生成粗略的 3D 模型并修改 cif 文件以使其与 MODELLER 兼容
- hhsuitedb.py:使用预过滤、打包的 MSA/HMM 和索引文件构建 HHsuite 数据库
- splitfasta.pl : 将一个多序列 FASTA 文件拆分成多个单序列文件
- renumberpdb.pl:生成 PDB 文件,索引重新编号以匹配输入序列索引
- HHPaths.pm:带有 PDB、BLAST、PSIPRED 等路径的配置文件。
- mergeali.pl:根据种子序列的 MSA 以 A3M 格式合并 MSA
- pdb2fasta.pl:从全局 pdb 文件的 SEQRES 记录生成 FASTA 序列文件
- cif2fasta.py:从 globbed cif 文件的 entity_poly 的 pdbx_seq_one_letter_code 条目生成 FASTA 序列
- pdbfilter.pl:从 pdb2fasta.pl 输出中生成一组具有代表性的 PDB/SCOP 序列
- pdbfilter.py:从 cif2fasta.py 输出生成一组具有代表性的 PDB/SCOP 序列
调用不带参数或带-h选项的程序以获得更详细的解释。
使用 HHsearch 和 HHblits 搜索 HMM 的数据库
我们将使用HHsuite 子目录query.a3m中data/的 MSA 作为示例查询。要在 scop70_1.75数据库中搜索与查询序列或 MSA in 同源的序列query.a3m,请键入
hhsearch -cpu 4 -i data/query.a3m -d databases/scop70_1.75 -o data/query.hhr
该数据库scop70_1.75可以从我们的下载服务器获得。如果输入文件是一个 MSA 或单个序列,HHsearch 会从中计算一个 HMM,然后 scop70_1.75使用 Viterbi 算法将此查询 HMM 与数据库中的所有 HMM 对齐。搜索后,最重要的 HMM 使用更准确的最大精度 (MAC) 算法重新排列。在重新对齐阶段之后,由汇总命中列表和成对查询模板对齐组成的完整搜索结果将写入输出文件data/query.hhr,.hhr 结果文件格式被设计为人类可读且易于解析。
该-cpu 4选项告诉 HHsearch 启动四个 OpenMP 线程以进行搜索和重新对齐。这通常会使具有四个或更多内核的计算机的执行速度提高近四倍。由于线程的管理成本可以忽略不计。
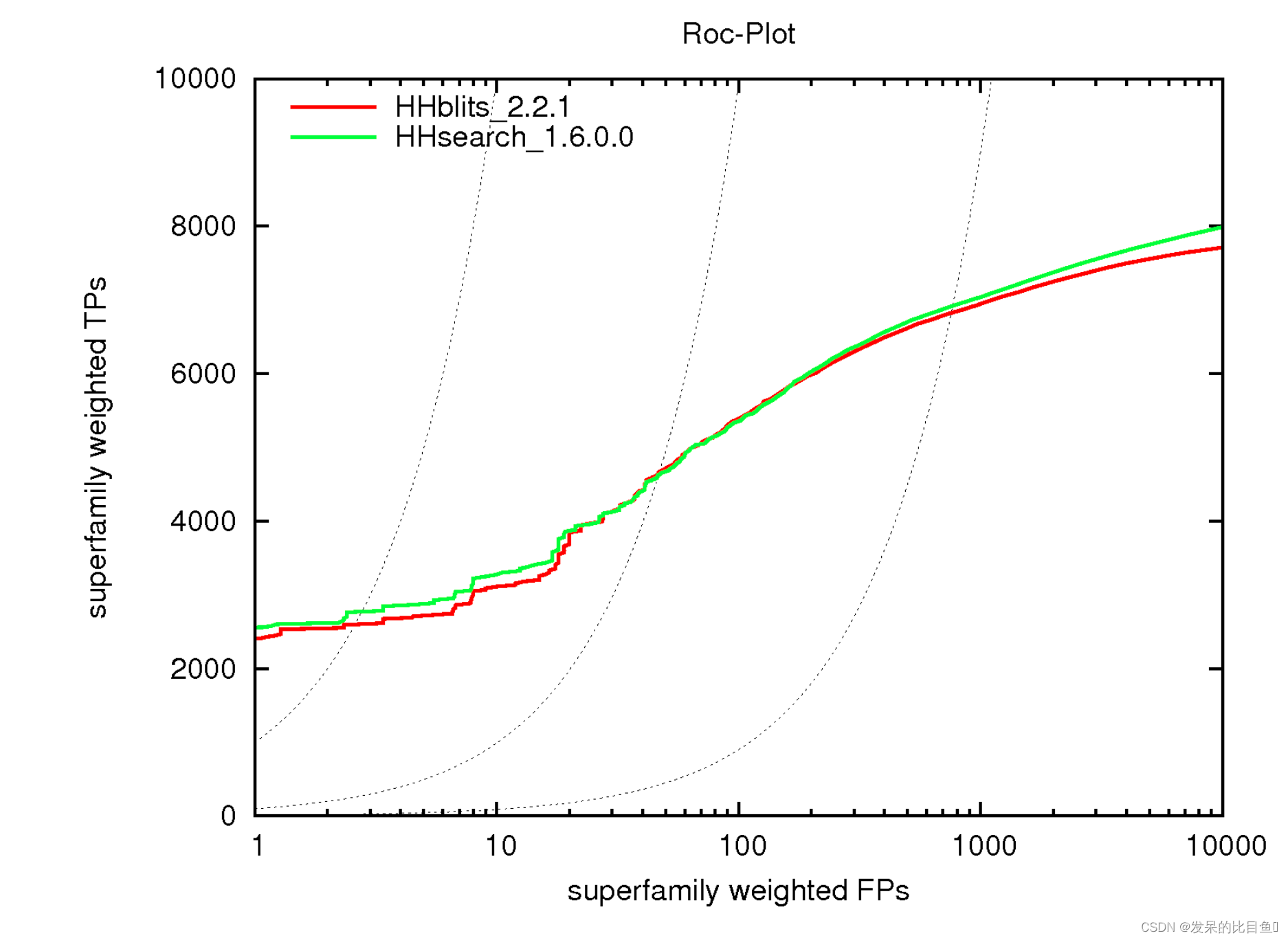
HHblits 工具的使用方式与 HHsearch 大致相同。它采用相同的输入数据并生成与 HHsearch 格式相同的结果文件。大多数 HHsearch 选项也适用于 HHblits,它具有与迭代搜索的扩展功能相关的附加选项。由于其快速的预过滤器,HHblits 的运行速度比 HHsearch 快 30 到 3000 倍,但灵敏度仅降低了几个百分点。
此处使用 HHblits 而不是 HHsearch 执行与上述相同的搜索:
hhblits -cpu 4 -i data/query.a3m -d databases/scop70_1.75 -o data/query.hhr -n 1
HHblits首先用它的快速预过滤器扫描scop70_1.75_cs219.ffdata中的列状态序列。列状态序列通过预过滤器的HMM从打包文件scop70_1.75_hhm.ffdata中读取(使用索引文件scop70_1.75_hhm.ffindex),并使用缓慢的Viterbi HMM-HMM对齐算法将其对齐到从query.a3m生成的查询HMM。搜索结果被写入默认输出文件查询。选项-n 1告诉HHblits执行一次搜索迭代。(默认是2次迭代)
使用HHblits生成多个序列对齐
为一个序列或query.a3m中的初始MSA生成一个MSA,要搜索的数据库应该覆盖整个序列空间,如Uniclust30。选项-oa3m <msa_file>告诉HHblits从中生成一个输出MSA, -n 1指定一个单一的搜索迭代。
hhblits -cpu 4 -i data/query.seq -d databases/uniclust30_2018_08/uniclust30_2018_08 -oa3m query.a3m -n 1
在搜索结束时,HHblits从包含msa的打包数据库文件中读取e值低于阈值的属于hmm的序列。可以使用-e <E-value>选项指定加入MSA的E-value阈值。搜索后,query.a3m将包含a3m格式的MSA。
我们可以进行第二次搜索迭代,从前一次搜索的MSA开始,以添加更多序列。由于在前面的搜索之后生成的MSA比query.seq中的单个序列包含更多的信息,因此使用这个MSA进行搜索可能会导致更多的同源数据库匹配
hhblits -cpu 4 -i query.a3m -d databases/uniclust30_2018_08/uniclust30_2018_08 -oa3m query.a3m -n 1
相反,我们可以直接从query.seq开始执行两次搜索迭代
hhblits -cpu 4 -i data/query.seq -d databases/uniclust30_2018_08/uniclust30_2018_08 -oa3m query.a3m -n 2
看为什么我执行2与1+1 HHblits迭代得到不同的结果?为了解释为什么结果会略有不同。
在实践中,建议使用1到4次迭代来构建msa,这取决于一边的可靠性和特异性和另一边的远程同源序列的敏感性之间的权衡。进行的搜索迭代越多,非同源序列或序列段进入MSA并在后续迭代中招募更多同类的风险就越高。这在搜索含有短重复序列、氨基酸组成偏序区域和具有多个结构域(尽管不太明显)的序列时尤其有问题。幸运的是,由于hhblits的迭代次数更少,其更健壮的最大精度对齐算法,以及其HMM-HMM对齐的更高精度,与PSI-BLAST相比,这个问题在hhblits中不太明显。
参数-mact(最大精度阈值)允许您在灵敏度和精度之间进行取舍。使用较低的mact值(例如-mact 0.01)非常敏感,但不那么精确的对齐会生成,而使用较高的mact值(例如-mact 0.9)的搜索会生成较短但非常精确的对齐。HHblits中-mact的默认值是0.35。
为了避免不必要的大型和多样化的MSA,当查询MSA的多样性作为有效序列的数量(N eff)增长超过阈值10.0时,HHblits停止迭代。这个阈值可以通过--neffmax <float>选项进行修改。有关如何在HH-Suite中计算有效序列的数量,请参见HHSearch/HHBLITS模型格式。
为了避免最终MSA变得不必要地大,默认情况下,将与查询MSA合并到结果MSA中的数据库MSA将使用活动筛选选项进行筛选(默认情况下-id 90和-diff1000)。-all选项关闭对结果MSA的过滤。如果您想获得非常相似的Uniclust30集群中的所有序列,请使用此选项
hhblits -cpu 4 -i data/query.seq -d databases/uniclust30_2018_08/uniclust30_2018_08 -oa3m query.a3m -all
A3M格式使用小写字母标记插入,使用大写字母指定匹配和删除列(请参阅多种序列对齐格式),允许您省略对齐以插入列的间隙。因此,A3M格式使用的大对齐空间比FASTA少得多,但在人眼看来是不对齐的。使用reformat.pl脚本将query.a3m重新格式化为其他格式,例如将MSA重新格式化为Clustal和FASTA格式,键入
reformat.pl a3m clu query.a3m query.clu
reformat.pl a3m fas query.a3m query.fas
接下来,要向MSA添加二级结构信息,我们调用脚本addss.pl。对于addss.pl运行,您必须确保正确填写$HHLIB/scripts/HHPaths.pm中BLAST和PSIPRED的路径:
addss.pl query.a3m
当序列的名称中的第一个单词是SCOP或PDB标识符时,脚本也会试着添加DSSP状态。打开query.a3m文件,签出已添加到MSA的两行。
虽然二级结构评分确实在一定程度上改善了同源性检测,但这一步非常耗时,而且由于工具的老版本是硬编码的,目前很难设置。我们建议跳过此步骤。
现在您可以从这个MSA生成一个隐马尔可夫模型(HMM)
hhmake -i query.a3m -o query.hhm
默认输出文件是query.hhm. 默认情况下,-M first将使用该选项。这意味着在查询序列中包含残基的那些 MSA 列将被分配到匹配/删除状态,其他列将被分配到插入状态。(查询序列是第一个不包含二级结构信息的序列。)或者,您可能希望通过输入-M 50来应用50%间隙规则,这将只将那些包含超过50%间隙的列分配给Insert状态。这-M first如果您的比对可以最好地被视为种子序列加上比对的同源物以通过进化信息加强它,则该选项是有意义的。在我们的 HMM 数据库的 SCOP 和 PDB 版本中就是这种情况,因为这里的 MSA 是围绕单个种子序列(具有已知结构的序列)构建的。相反,当您的比对代表整个同系物家族并且没有特别的序列时,最好使用 50% 间隙规则。例如,Pfam 或 SMART MSA 就是这种情况。尽管很简单,但 50% 的差距规则已被证明在实践中表现良好。
当调用hhmake时,您还可以应用几个过滤器,例如最大双向序列标识(-id <int>),查询序列的最小序列标识(-qid <int>),或查询的最小覆盖率(-cov <int>)。
示例:使用 HHblits 和 MODELLER 进行比较蛋白质结构建模
三维 (3D) 结构极大地促进了蛋白质的功能表征。然而,对于许多蛋白质来说,没有可用的实验结构,因此,与已知蛋白质结构的比较建模可能会提供有用的见解。在该方法中,基于与一种或多种已知结构的蛋白质(模板)的比对来预测给定蛋白质序列(目标)的 3D 结构。在下文中,我们将演示如何使用 HHblits 和 PDB70 数据库为未解析的蛋白质创建比对。然后,我们将 HHblits 的搜索结果转换为使用 MODELLER (v9.16) 构建比较模型。
1999年,吴等人。报道了来自阴道毛滴虫 (TvLDH)的乳酸脱氢酶基因的基因组序列和进化分析。令人惊讶的是,相应的蛋白质序列与同一生物体的苹果酸脱氢酶 (TvMDH) 最相似,这意味着 TvLDH 通过趋同进化源自 TvMDH。与此同时,TvLDH 的结构已被解析,然而,出于教学目的,假设该蛋白质没有 3D 结构。
首先,我们从 GeneBank 获得 TvLDH 的蛋白质序列(登录号 AF060233.1)。我们首先将蛋白质序列复制粘贴到一个名为query.seq. 该文件的内容应该与此类似。
>TvLDH
MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLAGFVATTDP
KAAFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGNPDNTNCEIAMLHAKNL
KPENFSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLTQATFTKEGKTQKVVDVLDHDYVFD
TFFKKIGHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTAPGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVD
KEGKIHVVEGFKVNDWLREKLDFTEKDLFHEKEIALNHLAQ
如果我们使用 MSA 而不是单个序列,则通过针对 PDB70 查询 TvLDH 序列获得的搜索结果将明显更好。为此,我们首先针对覆盖整个蛋白质序列空间的 Uniclust30 数据库查询 TvLDH 的蛋白质序列。通过执行
hhblits -i query.seq -d databases/uniclust30_2018_08/uniclust30_2018_08 -oa3m query.a3m -cpu 4 -n 1
我们获得了一个 a3m 格式的 MSA,其中包含几个类似于 TvLDH 的序列。现在我们可以使用query.a3m并搜索 PDB70 数据库来寻找相似的蛋白质结构。
hhblits -i query.a3m -o results.hhr -d databases/pdb70 -cpu 4 -n 1
请注意,我们现在输出的是 hhr 文件results.hhr而不是 a3m 文件。在我们将搜索结果转换为 MODELLER 可读的格式之前,让我们快速检查一下results.hhr.
Query TvLDH
Match_columns 333
No_of_seqs 2547 out of 8557
Neff 11.6151
Searched_HMMs 1566
Date Tue Aug 16 11:35:02 2016
Command hhblits -i query.a3m -o results.hhr -d pdb70 -cpu 32 -n 2
No Hit Prob E-value P-value Score SS Cols Query HMM Template HMM
1 7MDH_C MALATE DEHYDROGENASE; C 100.0 1.5E-39 1.3E-43 270.1 0.0 326 3-333 31-358 (375)
2 4UUL_A L-LACTATE DEHYDROGENASE 100.0 1.8E-35 1.6E-39 244.5 0.0 332 1-332 1-332 (341)
3 4UUP_A MALATE DEHYDROGENASE (E 100.0 2.2E-35 1.9E-39 243.8 0.0 332 1-332 1-332 (341)
4 4UUM_B L-LACTATE DEHYDROGENASE 100.0 5.7E-35 5.1E-39 241.5 0.0 333 1-333 1-333 (341)
5 1CIV_A NADP-MALATE DEHYDROGENA 100.0 1.6E-34 1.5E-38 241.3 0.0 326 2-332 40-367 (385)
6 1Y7T_A Malate dehydrogenase(E. 100.0 3.4E-34 3.1E-38 235.3 0.0 324 1-331 1-325 (327)
7 4I1I_A Malate dehydrogenase (E 100.0 3.8E-34 3.4E-38 236.4 0.0 319 2-327 22-343 (345)
8 1BMD_A MALATE DEHYDROGENASE (E 99.9 5.9E-34 5.3E-38 233.9 0.0 324 1-331 1-325 (327)
9 4H7P_B Malate dehydrogenase (E 99.9 9.9E-34 8.9E-38 233.9 0.0 319 2-327 22-343 (345)
10 2EWD_B lactate dehydrogenase, 99.9 1.1E-33 9.5E-38 231.1 0.0 306 1-328 1-314 (317)
(...)
我们发现有几个模板与我们的查询具有高度相似性。有趣的是,E 值得分最高的命中也是苹果酸脱氢酶。我们将使用这种结构作为我们比较模型的基础。为了构建模型,我们首先必须获得模板结构。我们可以通过输入以下命令来获得 7MDH
mkdir templates
cd templates
wget http://files.rcsb.org/download/7MDH.cif
cd ..
要将我们的搜索结果results.hhr转换为 MODELLER 可读的对齐方式,我们使用hhmakemodel.py.
python3 hhmakemodel.py results.hhr templates/ TvLDH.pir ./ -m 1
该脚本采用四个位置参数:hhr 格式的结果文件、包含所有 cif 格式模板的文件夹的路径、输出 pir 文件以及应写入已处理 cif 文件的文件夹。该-m标志告诉hhmakemodel.py仅在 pir 对齐中包含第一个命中。pir 文件连同处理过的 cif 可用作 MODELLER 的输入(请参阅 MODELLER 文档以获取更多帮助)。
目视检查 MSA 是否有损坏的区域
诸如 PSI-BLAST 和 HHblits 等迭代搜索方法可以生成包含非同源序列延伸甚至大部分非同源序列的比对。造成这种情况的原因几乎总是同源比对过度延伸到非同源区域。这被称为同源过度扩展在 。这种效应尤其发生在多结构域和重复蛋白质中。搜索结果中的单个过度延伸比对导致更多此类序列包含在下一次迭代的配置文件中 - 通常与第一个有问题的序列同源,但具有更长的非同源延伸。因此,在三次或更多次迭代之后,产生的 MSA 的大部分可能与原始查询序列非同源。与 PSI-BLAST 相比,HHblits 中同源过度延伸的风险大大降低,因为 HHblits 通常需要更少的迭代,并且因为 HHblits 使用最大精度对齐算法(请参阅最大精度对齐算法做什么?),与大多数其他程序(包括 PSI-BLAST)使用的 Smith-Waterman/Viterbi 算法相比,它更不容易过度扩展对齐。尽管如此,在重要的情况下,还是有必要目视检查查询的 MSA 和匹配的数据库蛋白质是否存在包含非同源序列延伸的损坏区域。
目视检查 MSA 的推荐程序如下。我们首先将 MSA 简化为一小组序列,这些序列仍然可以放入对齐查看器的单个窗口中:
hhfilter -i query.a3m -o query.fil.a3m -diff 30
该选项-diff使 hhfilter 选择一组至少 30 个最能代表 MSA 完整多样性的序列的代表性集合。因此,通常保留包含非同源片段的序列,因为它们往往与主序列簇最不相似。
接下来,我们删除-r与 MSA 中第一个主序列相关的所有插入(选项)。生成的 MSA 有时称为主从对齐:
reformat.pl -r query.fil.a3m query.fil.fas
这种对齐现在很容易在任何允许根据其物理化学性质对残基着色的查看器中检查有问题的区域。我们可以推荐alnedit 或jalview,例如:
java -jar alnedit.jar query.fil.fas .
构建自定义数据库
可以使用我们用于构建标准 HH-suite 数据库的相同工具构建自定义 HH-suite 数据库(Uniclust30 除外)。一个示例应用是在生物体的所有蛋白质中搜索同源物。要从一组序列构建您自己的 HH-suite 数据库,您首先需要为该组中的每个序列生成一个具有预测二级结构的 MSA。
以下调用假定您已经编译了支持 MPI 的 HH 套件。您可以替换hhblits_mpi为hhblits_omp、ffindex_apply_mpiwithffindex_apply和cstranslate_mpiwithcstranslate -f以使用 OpenMP(单计算节点)并行化。
创建与 HHblits 兼容的 MSA 的数据库
从序列开始
首先,我们将输入的 FASTA 文件转换为 FFindex 数据库 ffindex_from_fasta:
ffindex_from_fasta -s <db>_fas.ff{data,index} <db.fas>
现在,要为 中的每个序列构建一个带有 HHblits 的 MSA <db>_fas.ff{data,index},运行
mpirun -np <number_threads> \
hhblits_mpi -i <db>_fas -d <path_to/uniclust30> -oa3m <db>_a3m_wo_ss -n 2 -cpu 1 -v 0
MSA 被写入 ffindex <db>_a3m_wo_ss.ff{data,index}。为确保一切顺利,请检查中的行数 <db>_a3m.ffindex是否与中的行数相同<db>_fas.ffindex。
-n <int>HHblits 搜索迭代的次数和 HHblits 的 HMM 包含 E 值阈值可以使用和-e <float>选项从它们的默认值(分别为 2 和 0.01)更改。更高数量的迭代例如-n 3将导致发现远程同源关系的灵敏度非常高,但同源蛋白质之间的排名通常不会反映它们与查询的关系程度。原因是任何高于查询和数据库 MSA 中序列相似度的相似度都无法解析。
从 MSA 开始
如果您已经有对齐的 FASTA 格式的 MSA,请将它们首先放在不包含任何其他文件的单个文件夹中,以创建单个 FFindex 数据库:
cd msa/
ffindex_build -s ../<db>_msa.ff{data,index} .
cd ..
现在您可以使用hhconsensus添加一致序列并将格式转换为更节省空间的 A3M 格式:
OMP_NUM_THREADS=1 mpirun -np <number_threads> ffindex_apply_mpi <db>_msa.ff{data,index} \
-i <db>_a3m_wo_ss.ffindex -d <db>_a3m_wo_ss.ffdata \
-- hhconsensus -M 50 -maxres 65535 -i stdin -oa3m stdout -v 0
rm <db>_msa.ff{data,index}
标注二级结构信息(可选)
现在,将 PSIPRED 预测的二级结构和可能的 DSSP 二级结构注释添加到所有 MSA:
虽然二级结构评分确实改善了同源性检测,但此步骤非常耗时,并且由于旧版本的工具硬编码,目前很难设置。我们建议跳过此步骤。将 _a3m_wo_ss.ff{data,index} 重命名为 _a3m.ff{data,index} 以继续下一步。
mpirun -np <number_threads> ffindex_apply_mpi <db>_a3m_wo_ss.ff{data,index} \
-i <db>_a3m.ffindex -d <db>_a3m.ffdata -- addss.pl -v 0 stdin stdout
rm <db>_a3m_wo_ss.ff{data,index}
计算隐马尔可夫模型
我们还需要为每个 MSA 生成一个 HHM 模型:
mpirun -np <number_threads> ffindex_apply_mpi <db>_a3m.ff{data,index} \\
-i <db>_hhm.ffindex -d <db>_hhm.ffdata -- hhmake -i stdin -o stdout -v 0
计算用于预过滤的上下文状态
为了构建包含列状态序列的 FFindex 数据库,用于对我们运行的每个 a3m 进行预过滤:
mpirun -np <number_threads> cstranslate_mpi -f -x 0.3 -c 4 -I a3m -i <db>_a3m -o <db>_cs219
注意参数。遗漏任何内容可能会导致数据库看起来表面上正确,但性能却很差。
在版本 3.2 之前,您需要指定-b参数来cstranslate生成与 HHblits3 兼容的数据库,如果没有它,您将生成一个仍然可以后处理以供 HHblits2.x 读取的数据库。我们已经放弃了对 HHblits2.x 和-b参数的支持。
把所有东西放在一起
接下来我们要重新排序hmm和a3m数据库中的条目。为此,我们根据 MSA 中的列数(即共有序列的序列长度)对数据文件中的文件进行排序。可以在 的第三行中检索列数_cs219.ffindex。
sort -k3 -n -r <db>_cs219.ffindex | cut -f1 > sorting.dat
ffindex_order sorting.dat <db>_hhm.ff{data,index} <db>_hhm_ordered.ff{data,index}
mv <db>_hhm_ordered.ffindex <db>_hhm.ffindex
mv <db>_hhm_ordered.ffdata <db>_hhm.ffdata
ffindex_order sorting.dat <db>_a3m.ff{data,index} <db>_a3m_ordered.ff{data,index}
mv <db>_a3m_ordered.ffindex <db>_a3m.ffindex
mv <db>_a3m_ordered.ffdata <db>_a3m.ffdata
示例:从 PDB 构建数据库
为了在 PDB 中进行有效的序列搜索,我们提供了一个预编译的 PDB70 数据库,其中包含以 70% 序列同一性聚集的 PDB 序列。但是,要找到更多种类的 PDB 模板,可能需要具有更多冗余的更大数据库。在本教程中,我们概述了构建自定义 PDB 数据库所需的步骤。有关用于构建此数据库的实际脚本,请参阅PDB70 GitHub存储库。
首先,通过执行以 文件格式(这是文件格式的继承者)从RSCB下载整个 PDB 数据库 cifpdb
rsync --progress -rlpt -v -z --port=33444 rsync.wwpdb.org::ftp/data/structures/divided/mmCIF <cif_dir>
并将文件解压缩到一个目录<all_cifs>中。然后, cif2fasta.py通过键入运行
python3 cif2fasta.py -i <all_cifs> -o pdb100.fas -c <num_cores> -p pdb_filter.dat
该脚本扫描文件夹<all cifs>,查找后缀为*.cif的文件,并将entity_poly表中pdbx_seq_one_letter_code每个序列及其相关链标识符写入fasta文件pdb100.fas。通过指定可选的-p标志,cif2fast .py创建一个额外的文件pdb filter.dat, pdbfilter.py在后面的步骤中需要这个文件。请注意,默认情况下,cif2fasta.py删除了比30个残基短的序列和/或仅包含残基’x’。
如果希望彻底搜索PDB,请跳过以下步骤,并继续学习构建自定义数据库一节中描述的说明。然而,为了提高数据库搜索的速度,例如在资源有限的系统上,您可以通过使用MMseqs2聚类来减少序列的数量,并使用pdbfilter.py选择具有代表性的序列。将pdb100的序列聚类。fas在序列标识为X和覆盖范围为Y处使用这些选项运行MMseqs2。
mmseqs createdb pdb100.fas <clu_dir>/pdb100
mmseqs cluster <clu_dir>/pdb100 <clu_dir>/pdbXX_clu /tmp/clustering -c Y --min-seq-id X
mmseqs createtsv <clu_dir>/pdb100 <clu_dir>/pdb100 <clu_dir>/pdbXX_clu <clu_dir>/pdbXX_clu.tsv
MMSEQS2产生一个选项卡分离的文件pdbxx_clu.tsv,其中包含所有序列的群集分配。代表性序列是由pdbfilter.py选择的,它通过识别具有最高分辨率A(最大的无R-FORATE值或最大的“完整性”)来为每个群集选择多达三个序列。
我们通过将原子部分中的残基数除以entity_poly表的pdbx_seq_one_letter_code输入中声明的残基总数来计算蛋白质结构的完整性。
python3 pdbfilter.py pdb100.fas pdbXX_clu.tsv pdb_filter.dat pdbXX.fas -i pdb70_to_include.dat -r pdb70_to_remove.dat
pdbfilter.py获取原始fasta文件(pdb100.fas)和注释文件pdb_filter.dat,这两者都是由cif2fasta.py创建的,以及MMseqs2(pdb70_clu.tsv)的群集分配作为输入,并输出最终的pdbxx.fas。使用这个fasta文件来完成数据库的创建(请参阅构建定制数据库一节)。
经常问的问题
略。。。
HHsearch/HHblits 输出:命中列表和成对对齐
摘要命中列表
让我们用人类 PIP49/FAM69B 蛋白进行搜索,我们在使用 HHblits生成多序列比对query.a3m中生成了一个 MSA,其中包含两次 HHblits 迭代 :
Query sp|Q5VUD6|FA69B_HUMAN Protein FAM69B OS=Homo sapiens GN=FAM69B PE=2 SV=3
Match_columns 431
No_of_seqs 149 out of 272
Neff 5.2
Searched_HMMs 13730
Date Wed Jan 4 17:44:24 2012
Command hhsearch -i query.a3m -d /cluster/user/soeding/databases/scop -cpu 18
No Hit Prob E-value P-value Score SS Cols Query HMM Template HMM
1 d1qpca_ d.144.1.7 (A:) Lymphoc 99.7 4.5E-17 3.2E-21 154.3 10.2 99 203-320 56-157 (272)
2 d1jpaa_ d.144.1.7 (A:) ephb2 r 99.7 4.3E-17 3.1E-21 156.8 8.8 99 203-321 75-177 (299)
3 d1uwha_ d.144.1.7 (A:) B-Raf k 99.7 5.1E-17 3.7E-21 154.8 7.7 100 203-322 52-154 (276)
4 d1opja_ d.144.1.7 (A:) Abelson 99.7 6.2E-17 4.5E-21 154.8 8.3 100 203-321 61-164 (287)
5 d1mp8a_ d.144.1.7 (A:) Focal a 99.6 9.9E-17 7.2E-21 151.3 8.6 100 203-322 56-158 (273)
6 d1sm2a_ d.144.1.7 (A:) Tyrosin 99.6 1.2E-16 8.8E-21 150.3 8.8 99 203-321 48-150 (263)
7 d1u59a_ d.144.1.7 (A:) Tyrosin 99.6 2.4E-16 1.7E-20 150.9 9.5 99 203-321 57-158 (285)
8 d1xbba_ d.144.1.7 (A:) Tyrosin 99.6 2.2E-16 1.6E-20 150.2 8.6 97 203-320 56-155 (277)
9 d1vjya_ d.144.1.7 (A:) Type I 99.6 2.6E-16 1.9E-20 151.3 8.8 98 204-320 46-156 (303)
10 d1mqba_ d.144.1.7 (A:) epha2 r 99.6 4.4E-16 3.2E-20 148.0 8.7 193 203-422 57-272 (283)
...
64 d1j7la_ d.144.1.6 (A:) Type II 97.3 0.00014 1E-08 65.0 6.3 33 292-324 184-216 (263)
65 d1nd4a_ d.144.1.6 (A:) Aminogl 96.7 0.0012 8.5E-08 58.5 6.6 31 292-322 176-206 (255)
66 d1nw1a_ d.144.1.8 (A:) Choline 96.6 0.0011 7.8E-08 63.9 5.8 37 203-239 92-128 (395)
67 d2pula1 d.144.1.6 (A:5-396) Me 95.6 0.0071 5.2E-07 58.3 6.4 32 290-322 222-253 (392)
68 d1a4pa_ a.39.1.2 (A:) Calcycli 91.7 0.12 8.9E-06 40.0 5.4 62 140-202 18-80 (92)
69 d1ksoa_ a.39.1.2 (A:) Calcycli 91.2 0.17 1.2E-05 39.5 5.8 56 147-203 28-83 (93)
70 d1e8aa_ a.39.1.2 (A:) Calcycli 90.5 0.23 1.7E-05 38.3 6.0 56 147-203 27-82 (87)
...
175 d1qxpa2 a.39.1.8 (A:515-702) C 23.7 29 0.0021 28.8 3.8 49 137-197 69-118 (188)
176 d1tuza_ a.39.1.7 (A:) Diacylgl 23.5 55 0.004 25.3 5.3 55 143-201 44-106 (118)
177 d1ggwa_ a.39.1.5 (A:) Cdc4p {F 23.1 26 0.0019 27.0 3.2 66 129-197 35-101 (140)
178 d1topa_ a.39.1.5 (A:) Troponin 22.8 72 0.0052 24.5 6.0 58 140-199 65-123 (162)
179 d1otfa_ d.80.1.1 (A:) 4-oxaloc 22.5 66 0.0048 21.5 5.0 40 267-306 12-53 (59)
180 d1oqpa_ a.39.1.5 (A:) Caltract 22.2 32 0.0023 24.0 3.2 32 165-197 3-34 (77)
181 d1df0a1 a.39.1.8 (A:515-700) C 21.7 43 0.0032 27.0 4.5 51 137-199 67-118 (186)
182 d1zfsa1 a.39.1.2 (A:1-93) Calc 21.1 41 0.003 24.6 3.8 30 170-199 8-38 (93)
183 d1snla_ a.39.1.7 (A:) Nucleobi 20.9 23 0.0016 26.2 2.2 24 174-197 18-41 (99)
写入文件的摘要命中列表显示数据库中的最佳命中,按真正肯定的概率排序(第 4 列:“概率”)。列的含义如下
No: 数据库匹配的索引。
Hit: 名称行的前 30 个字符。
Prob:模板为真阳性的概率。对于成为真阳性的概率,列中的二级结构得分与列SS中的原始得分一起被考虑在内Score。真阳性被定义为全局同源或它们至少部分同源,因此在结构上局部相似。更准确地说,后一个标准要求查询和命中之间的 MAXSUB 分数至少为 0.1。在几乎所有情况下,结构相似性都是由于查询和模板之间的全局 OR LOCAL 同源性。
E-value:E 值给出了平均误报数(“错误命中”),其得分高于扫描数据库时模板的得分。它是一种可靠性的衡量标准:接近 0 的 E 值表示非常可靠的命中,E 值 10 意味着预计在数据库中会找到大约 10 个错误命中,并且得分至少为这个好。请注意,计算 E 值和 P 值时没有考虑二级结构!
P-value:P值是E值除以数据库中的序列数。在成对 比较中,一个错误的命中至少会得到这么好的分数。
Score:原始分数由 Viterbi HMM-HMM 比对计算,不包括二级结构分数。它是对齐的配置文件列的相似性总和减去特定位置的间隙惩罚(以位为单位)。列相似性得分是原始 HHsearch 论文 (Soding, Bioinformatics 2005) 中描述的对数总和得分(以 2 为底)。间隙惩罚是状态转换概率的 log2,例如从匹配状态到插入或删除到匹配状态。
SS:二级结构得分。该分数告诉您 PSIPRED 预测的(3 态)或实际 DSSP 确定的(8 态)二级结构序列彼此之间的一致性程度。PSIPRED 置信度值用于评分,低置信度获得较少的统计权重。
Cols:HMM-HMM对齐中对齐的匹配列数。
Query HMM:来自查询 HMM 的对齐匹配状态的范围。
Template HMM:来自数据库/模板 HMM 的对齐匹配状态的范围,括号中是数据库 HMM 中匹配状态的数量。
HMM-HMM 成对比对
输出文件 d1bpya1.hhr 包含相同的命中列表以及成对的 HMM 对齐。一个例子是在这里:
No 68
>d1a4pa_ a.39.1.2 (A:) Calcyclin (S100) {Human (Homo sapiens), P11 s100a10, calpactin [TaxId: 9606]}
Probab=91.65 E-value=0.12 Score=40.00 Aligned_cols=62 Identities=16% Similarity=0.149 Sum_probs=42.0
Q ss_pred ccCCCCCCcHHHHHHHHHHHHHhhcccCccHHHHHHHHHhhhhccCCCCcCHHHHHHH-HHHHH
Q sp|Q5VUD6|FA69 140 FDKPTRGTSIKEFREMTLSFLKANLGDLPSLPALVGQVLLMADFNKDNRVSLAEAKSV-WALLQ 202 (431)
Q Consensus 140 ~d~p~~g~s~~eF~emv~~~i~~~lg~~~~l~~L~~~~~~~~d~nk~g~vs~~e~~sl-waLlq 202 (431)
||+..-..|.+||.+++.......++.+.+ ...+..++..+|.|+||+|++.|...+ ..|..
T Consensus 18 yd~ddG~is~~El~~~l~~~~~~~~~~~~~-~~~v~~~~~~~D~n~DG~I~F~EF~~li~~l~~ 80 (92)
T d1a4pa_ 18 FAGDKGYLTKEDLRVLMEKEFPGFLENQKD-PLAVDKIMKDLDQCRDGKVGFQSFFSLIAGLTI 80 (92)
T ss_dssp HHGGGCSBCHHHHHHHHHHHCHHHHHHSCC-TTHHHHHHHHHCTTSSSCBCHHHHHHHHHHHHH
T ss_pred HcCCCCEEcHHHHHHHHHHhccccccccCC-HHHHHHHHHHHhCCCCCCCcHHHHHHHHHHHHH
Confidence 444433449999999998876655554332 234566677899999999999997544 44443
该比对显示了嵌入在 PIP49/FAM69B 激酶结构域中的 EF 手。以 a 开头的第一行>包含模板/数据库 HMM 的名称和描述行。(我们使用“模板 HMM”和“匹配数据库 HMM”的同义词。)下一行总结了对齐的主要统计数据:查询和模板 HMM 同源的概率( Probab) E-value、Score从摘要命中列表中重复对齐的列。Identities给出查询的比对残基对和相同的模板主序列的百分比。这Similarity是来自查询和模板主序列的比对残基对之间替换分数的算术平均值。替换矩阵与用于计算数据库 HMM 的伪计数的矩阵相同,默认情况下为 Gonnet 矩阵。(可以使用-Blosom<XX>选项更改矩阵。)
该Sum_probs值是所有对齐的匹配状态对的后验概率之和。这些概率是通过 Forward-Backward 算法计算的。(它们由计算最终对齐的最大精度算法使用。)当模板 HMM 具有来自 DSSP 的二级结构注释时,该sum_probs值仅在模板具有有效 DSSP 状态而不是-符号的对齐对上运行。A-表示模板中缺少该残基的结构坐标。对于同源建模,这种对具有已知结构的模板的特殊处理使得sum_probs用于对模板进行排名的有用功能。
成对对齐由一个或多个具有以下行的块组成:
Q ss_dssp: the query secondary structure as determined by DSSP (when available)
Q ss_pred: the query secondary structure as predicted by PSIPRED (when available)
Q <Q_name>: the query master sequence
Q Consensus: the query alignment consensus sequence
如果 PSIPRED 置信度值介于 0.7 和 1.0 之间,则预测的二级结构状态以大写字母显示,对于较低的置信度值,它们以小写字母给出。使用选项 -ssconf,ss_conf可以将行添加到通过 0 到 9 之间的数字报告 PSIPRED 置信度值的路线中(如最高 1.5 的版本)。
共有序列使用大写字母表示保守的列,小写表示部分保守的列。非保守列用波浪号标记~。粗略地说,出现概率为 (>+ 60%) 的氨基酸(在添加伪计数之前)写为大写字母,概率为 (>= 40%) 的氨基酸写为小写字母,其中空格包含在分数计数。更准确地说,当间隙校正的氨基酸部分
p_i(a) * N_eff(i)/(N_eff + 1)
大于 0.6 (0.4) 大写(小写)字母用于表示氨基酸 a。这里,p_i(a)是第 i 列中 a 的发射概率,N_eff是整个多重比对中的有效序列数(1 到 20 之间),N_eff(i)是子比对中的有效序列数,由那些在第 i 列。这些百分比与列中间隙的比例大致成反比增加,因此只有半胱氨酸和 50% 间隙的列会得到一个小写字母。
中间的线显示查询和模板氨基酸分布之间的列分数。它为对齐质量提供了有价值的指示。
= : column score below -1.5
- : column score between -1.5 and -0.5
. : column score between -0.5 and +0.5
+ : column score between +0.5 and +1.5
| : column score above +1.5
一个列分数单位大约对应于 0.6 位。从柱刻线来看, D.n.DG.i…E两个螺旋之间的转弯处保守基序周围的良好对齐是显而易见的。通过对比,围绕间隙的对齐得分仅比每个残基的零好一点,因此不是很可靠。
在由以下几行组成的模板块之后,
T Consensus: the template alignment consensus sequence
T <T_name>: the template domain sequence
T ss_dssp: the template secondary structure as determined by DSSP (when available)
T ss_pred: the template secondary structure as predicted by PSIPRED (when available)
块中的最后一行(置信度)报告了成对查询模板对齐的可靠性。置信度值是从在 Forward-Backward 算法中计算的后验概率获得的。值 8 表示这对 HMM 列在 0.8 和 0.9 之间正确对齐的概率。The Confidence line is only displayed when the -realignoption is active.
文件格式
略。。。
命令行参数汇总
hhblits – 基于 HMM-HMM 的快速迭代序列搜索
HHblits 是一种敏感的通用迭代序列搜索工具,它通过 HMM 表示查询和数据库序列。您可以从单个查询序列、多序列比对 (MSA) 或 HMM 开始搜索 HHsuite 数据库。HHblits 打印出数据库 HMM/MSA 的排序列表,还可以通过将重要的数据库 HMM/MSA 合并到查询 MSA 上来生成 MSA。
该二进制文件hhblits_omp支持使用 OpenMP 对多个查询进行并行化。在这种情况下,输入需要是带有 MSA 的 ffindex 的基本名称。输出被视为输出 ffindices 的基本名称
假设 ffindex 数据库 /home/user/databases/scop_a3m.ff{data,index},对应的基本名称是/home/user/databases/scop_a3m
# a3m、a2m或FASTA格式的单序列或多序列对齐(MSA),或hhm格式的HMM

















 766
766

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









