






欢迎关注"生信修炼手册"!
haploview 是基于图形界面的软件,其界面设计良好,用法简单,是进行连锁不平衡分析的主流软件之一。
需要两个输入文件, 后缀分别为ped和info。ped文件保存的是样本的基因分型结果,这种格式在之前的文章中详细介绍过了;info文件保存的是SNP位点的ID和位置信息,内容如下
IGR1118a_1 274044
IGR1119a_1 274541
IGR1143a_1 286593第一列为SNP位点的ID, 第二列为SNP位点在基因组上的位置。
1. 导入输入文件
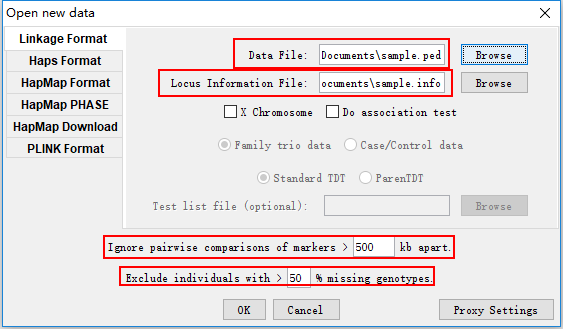
点击Linkage Format菜单,然后选择对应的输入文件。Data File 指定输入的ped文件,Locus Information File 指定输出的info文件,如果ped和info文件同名,在指定Data File的同时,程序会自动识别info文件;Ignore pairwise comparisons of markers 指定计算LD的范围,默认只对距离在500kb以内的SNP位点分析连锁不平衡,可以根据自己的需要进行调整,比如调整到1000Kb;Exclude individuals 对样本进行过滤,默认基因型缺失的比例大于50%的样本被剔除。当所有参数设置好之后,点击OK按钮即可。
2. 对输入的SNP位点进行过滤
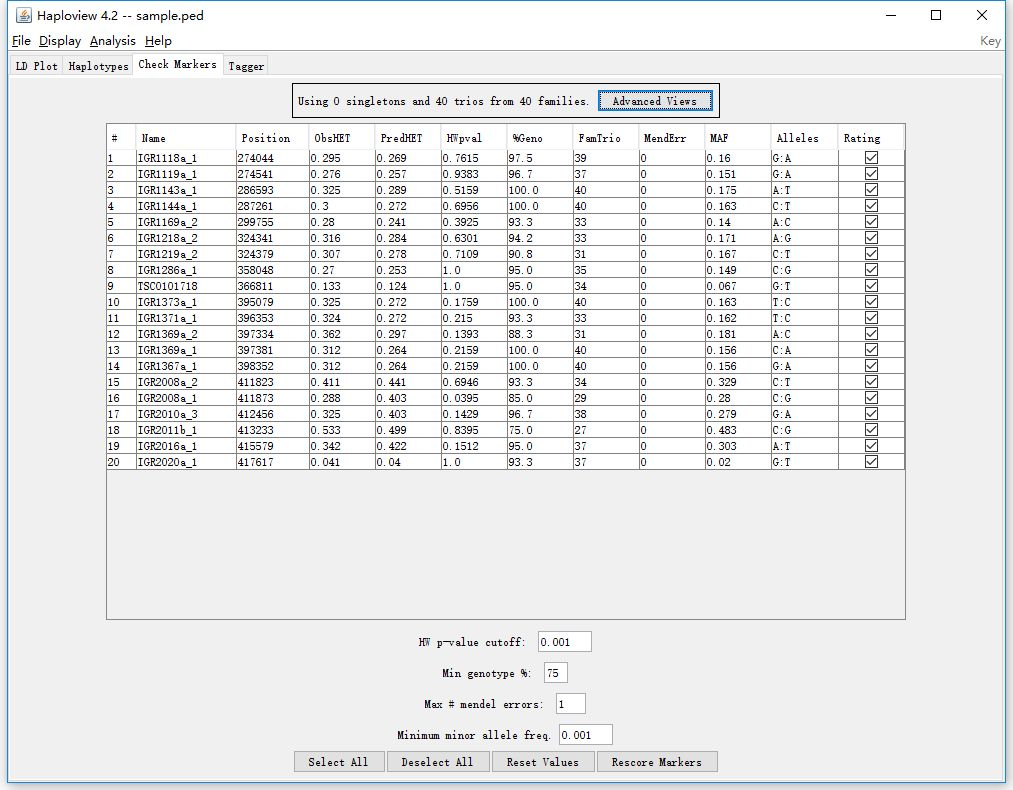
check markers可以对输入的SNP位点进行过滤, 可以根据HW pvalue, Min genotype, mendel error, MAF等阈值过滤。设置好相应的阈值之后,点击Rescore Markers按钮,就可以根据阈值过滤了,最后一列的Rating如果勾选上了,表示该SNP位点符合要求。
3. LD plot
点击LD plot 按钮,就可以看到如下所示的连锁不平衡的热图;每个格子代表了两个SNP位点之间的LD分析结果,颜色从白色到红色,代表连锁程度从低到高。右键点击格子,可以看到LD分析的详细结果。方框中的数值为D'值,为了美观,这里乘以了100。通过最上方的Display按钮,可以调整热图的外观。
相互之间高度连锁的SNP位点构成了haplotype block, 比如下图中的1-8构成了block1, 长度为84kb。

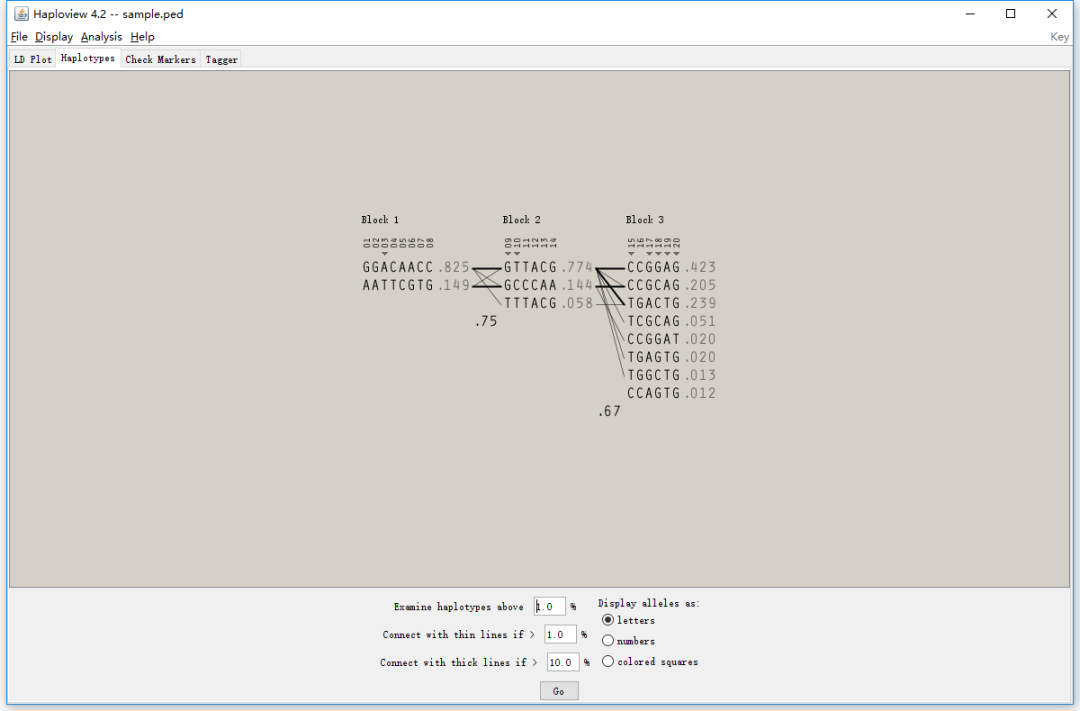
4. haplotypes
基于LD分析的结果,可以去定义一个haplotype block。 通过最上方的Analysis按钮,可以调整计算haplotype block的算法
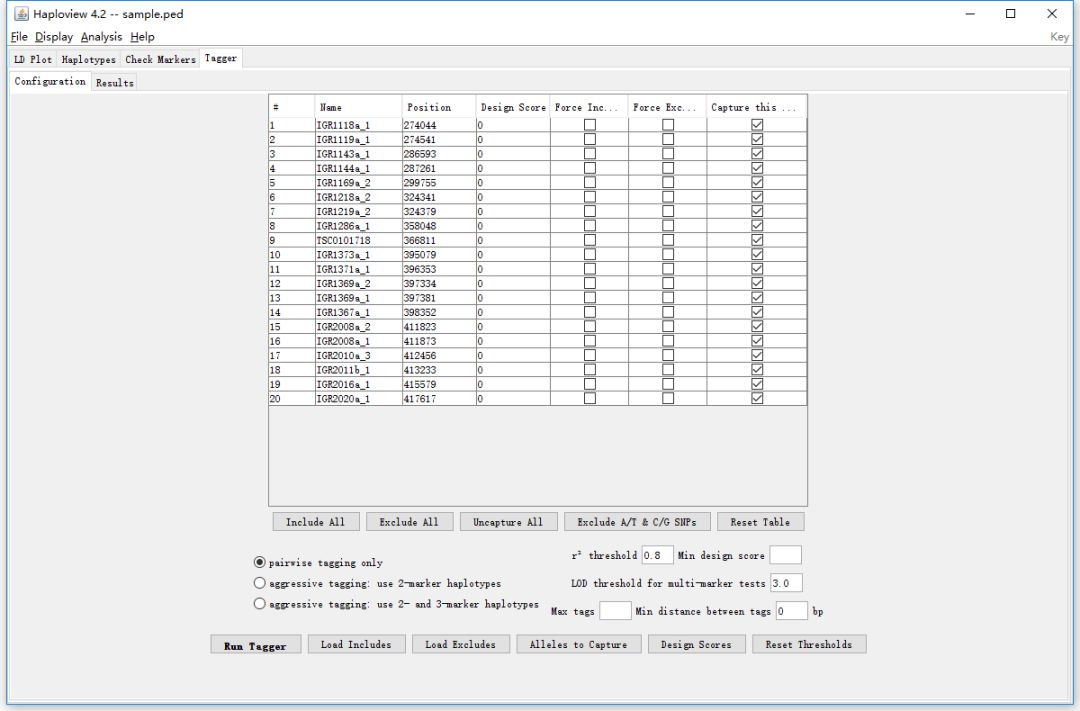
5. Tagger
Tagger按钮用于挑选tagSNPs, Configuration有两个用途,第一个是筛选SNP位点,通过勾选对应的单选框,可以指定想要进行分析的SNP位点;第二个用途是指定tagSNPs挑选的算法,默认是pairwise tagging only。
设置好之后,点击Run Ragger按钮,就可以了,结果界面如下
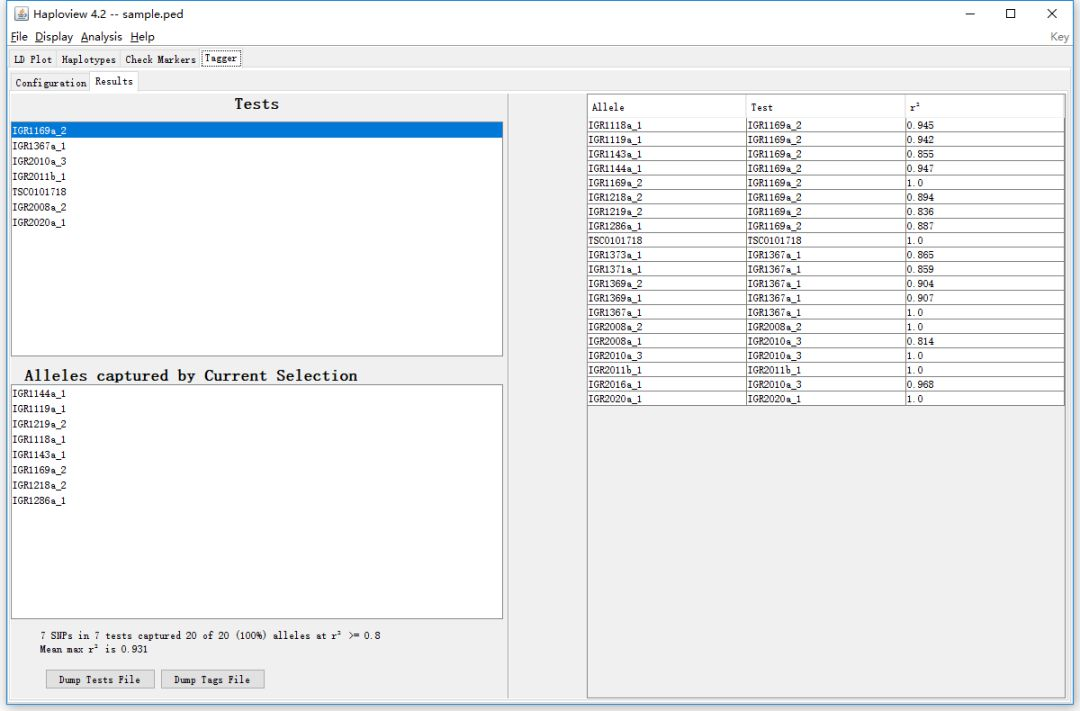
Test 框中显示的就是挑选出的tagSNPs, 鼠标左键单击每个SNP位点,在下方的Alleles captued by Current Selection 框中,会显示该tagSNP代表的其他SNP位点。右侧的表格展示了每个SNP位点对应的最佳的tagSNP。通过下方的Dump Tests File和Dump Tags File按钮可以导出结果。
扫描关注微信号,更多精彩内容等着你!


















 4993
4993

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









