










A haplotype-resolved genome provides insight into allele-specific expression in wild walnut (Juglans regia L.)
二倍体解析基因组为野生胡桃树(Juglans regia L.)的等位基因特异性表达提供了洞察

摘要
野生种质资源因其特殊的性状表现,对基因挖掘和分子育种至关重要。二倍体解析基因组是全面理解高度杂合物种亚基因组生物学的理想解决方案。本研究调查了中国新疆巩留县的一棵野生胡桃树的基因组,并利用PacBio高保真(HiFi)读取和Hi-C技术,为一个二倍体(hap1)生成了562.99 Mb(contig N50 = 34.10 Mb)的二倍体解析参考基因组,为另一个二倍体(hap2)生成了561.07 Mb(contig N50 = 33.91 Mb)的二倍体解析参考基因组。hap1的约527.20 Mb(93.64%)和hap2的约526.40 Mb(93.82%)被分配到16个拟染色体上。分别为hap1和hap2预测了41039和39744个蛋白编码基因模型。此外,两个二倍体基因组之间鉴定了123个结构变异(SVs)。最终鉴定出响应冷胁迫的等位基因特异性表达基因(ASEGs)。这些数据集可以用来研究亚基因组的进化,挖掘功能优良基因,以及探索与环境适应相关特定性状的转录基础。
背景与摘要
普通胡桃(Juglans regia L.)(2n=32)属于胡桃科的胡桃属,是重要的经济树种,也是第四大消费坚果的来源。普通胡桃的果实主要用于生产油和保健产品,因其脂质组成丰富并含有大量抗氧化剂,如酚类化合物、维生素和微量元素,这些成分在维护健康和预防疾病中具有潜在的益处。这些特性促进了多个国家如美国、伊朗、土耳其和中国的胡桃种植和生产。
胡桃很可能起源并被驯化于中亚,并在中国已有7000多年的历史。目前,中国被视为一个次级起源中心,拥有广泛的遗传多样性。野生胡桃在中国新疆巩留县的自然保护区内广泛分布。野生近缘种通常拥有在特定环境中对极端生物或非生物胁迫具有耐受性的优良等位基因,这些可以为遗传改良提供抗性资源。新疆野生胡桃显示出相对较高的抗逆性和良好的营养成分。特别是,野生胡桃的优良抗寒性可以保证其在巩留县长期极寒的冬季或晚春存活和繁衍。因此,该地区的野生胡桃是进行抗寒分子育种和鉴定抗寒应答机制的理想材料。然而,胡桃是一种非模式树种,大多数研究集中在受寒胡桃仁的品质或表型胡桃品种的抗寒性,很少关注抗寒的分子机制。
胡桃的草图基因组于2016年发布并在2018年得到改进。此后,通过长读长测序和Hi-C锚定,陆续发布了具有更高基因组连续性或染色体规模的参考基因组。此外,六种胡桃科(包括J. sigillata, J. cathayensis, J. mandshurica, J. hindsii, J. microcarpa 和 J. nigra)的基因组序列也已可用。然而,胡桃基因组在二倍体染色体之间具有高杂合性,当前的基因组是二倍体拼接。最近,在高度杂合生物中实现了二倍体定义的基因组组装,提供了关于杂合物种中等位基因变异的新见解和功能分化。
二倍体生物在其基因组中具有两个等位基因副本,但在特定个体中,这些等位基因不一定都活跃或以相同的水平活跃。由顺式作用的遗传变异或CHG甲基化引起的基因副本表达不均称称为等位基因特异性表达(ASE)。ASE首次在2002年的酵母中报道,并参与许多植物生物过程,如对轻微水分亏缺的反应以及通过部分至完全显性或过显性效应调节的杂种优势和染色质可及性的改变。近年来,得益于这一第三代测序时代的二倍体解析基因组组装,全基因组范围的ASE特征和相关基因已被很好地表征。在木本多年生植物苹果中,大约19%的表达基因是等位特异性的,许多ASE基因如编码ACC氧化酶和RIN样MADS-box转录因子的基因与果实品质相关。
本研究报告了一种高质量的野生胡桃二倍体解析参考基因组。首先,使用Illumina测序读数估计基因组大小、杂合性和重复序列的比例。然后,采用PacBio高保真(HiFi)长读数首先组装高连续性的拼接体,接着通过HiFi读数分相重组得到的二倍体。然后,通过Hi-C技术辅助确定了两个二倍体(hap1和hap2)的染色体规模。最终组装长度分别为hap1的562.99 Mb和hap2的561.07 Mb,拼接体N50值分别为33.91 Mb和22.40 Mb。大约93.64%的拼接体(527.2 Mb)被锚定到16个拟染色体上,93.82%(526.4 Mb)被锚定到hap2的16个拟染色体上。使用LTR组装指数(LAI v2.9.4)、BUSCO v5完整性、基因组共线性和分相切换错误评估二倍体基因组的完整性、连续性和分相准确性。我们为hap1和hap2预测了41039和39744个基因模型,分别由四个常规数据库功能注释34617和34562个基因。还分别为hap1和hap2鉴定了358869和358104个简单序列重复(SSRs)。突出显示了hap1和hap2之间的基因组变异,包括不同类型的结构变异(SVs)。最后,在冷处理后0小时、3小时、6小时、12小时和24小时分别鉴定了358、457、465、494和386个ASEGs。这种高质量的二倍体解析基因组组装将揭示亚基因组进化、分子遗传学和ASE调控机制,这些机制是野生胡桃适应极端寒冷环境的基础。
方法
植物材料准备、Illumina短读序列库构建和测序
从中国新疆巩留县自然保护区的一棵野生胡桃树上采集了大约20克的幼叶(东经82°16′28″,北纬43°20′52″)。野生胡桃的生境海拔范围从1241米到1670米,年平均温度为7.6°C,7月平均温度为19.7°C,1月平均温度为-9.9°C。在寒冷胁迫下,野生胡桃的表现优于幼苗胡桃。位置和外观见图1。然后,将叶子切成小片并迅速放入液氮中。使用Super Plant Genomic DNA DP360试剂盒(天根生物技术,北京,中国)提取新鲜叶片的基因组DNA,然后使用NanoDrop分光光度计(ND2000,赛默飞世尔科技,美国)测量DNA浓度,并通过在0.80%琼脂糖凝胶上进行电泳来监测质量。将高质量的DNA用Covaris S2仪器(Covaris,沃本,马萨诸塞州,美国)进行超声波碎片处理(350bp)。根据制造商的标准协议,使用TruSeq DNA样本制备试剂盒(Illumina,圣地亚哥,加利福尼亚,美国)准备350bp配对末端重测序库。使用安捷伦2100生物分析仪(安捷伦科技,帕洛阿尔托,加利福尼亚)和q-PCR来测量文库片段的大小和质量。为了辅助基因模型预测,从同一树的叶、根、芽和花组织混合样本中提取总RNA(约1克),使用RNeasy Plant Mini试剂盒(Qiagen,德国)按照制造商的说明书进行,并使用无RNase的DNase I消化试剂盒(Aidlab,中国)处理。使用1%琼脂糖凝胶监测RNA降解情况,使用NanoDrop 2000分光光度计(赛默飞世尔,威尔明顿,特拉华州,美国)测量分离RNA的纯度。在安捷伦2100生物分析仪上评估RNA的完整性,并使用TruSeq RNA文库制备试剂盒v.2(Illumina,圣地亚哥,加利福尼亚,美国)构建RNA文库。DNA和RNA文库均在Illumina NovaSeq 6000测序仪(Illumina,圣地亚哥,加利福尼亚,美国)上进行测序,按照制造商的推荐进行。原始读数经过过滤,通过去除接头序列、过滤含有>10%未识别核苷酸的读数和去除含有>50%低Phred质量分数(≤10)的碱基的读数,生成高质量的清洁读数。结果生成了31.47 Gb(约56倍覆盖度)的基因组调查和10 Gb的基因模型预测的高质量清洁读数(RNA测序数据)。

用于该基因组组装的野生胡桃的位置和外观。(a)采样野生胡桃的精确位置(红色箭头标示)。(b)采样野生胡桃的外观。PacBio HiFi 长读序列文库的生成和测序 根据 Mayjonade 的方法提取高完整性的基因组 DNA。根据 PacBio 的标准协议使用 HiFi 文库。简要地,将 10 微克高质量的 gDNA 用 Covaris g-TUBE 设备(Covaris,沃本,MA,美国)随机破碎,并进行 DNA 损伤修复和末端修复。然后连接哑铃形适配器。之后,产品经过外切酶消化,接着使用 BluePippin(Sege Science,美国)进行大小选择,筛选出 10 kb 至 30 kb 的片段。最终文库在 PacBio Sequel II 平台(循环一致性测序(CCS)模式)上进行测序,使用两个流动单元进行了 35 小时的测序(Adsen 生物技术公司,乌鲁木齐,中国)。最终,我们获得了大约 57.86 Gb 的 HiFi 读数(约 103 倍深度),CCS contig N50 为 15.91 kb,最大长度为 50.33 kb。
Hi-C 文库的准备和测序
由于活体样本可以提供体内的整合和实际的相互作用状态,因此收集野生胡桃的种子(用于 DNA 提取)并在室内发芽。四叶期的幼苗用于根据 Lieberman-Aiden 的方法准备 Hi-C 文库。简要地,新鲜叶子立即与甲醛交叉链接,然后用 Hind III(NEB)消化交联的 DNA。随后,这些酶消化片段的粘性末端被修复为生物素修饰的碱基。使用钝端近接连接的片段不断生成环形 DNA 分子,并将其破碎成 300-700 bp 的片段。最后,通过生物素珠子富集片段,在经过 Qbit 2.0(生命科技,卡尔斯巴德,加利福尼亚,美国)和 Agilent 2100 生物分析仪(Agilent Technologies,帕洛阿尔托,加利福尼亚,美国)进行文库质量控制后,该文库进行了一通道的 150 bp 配对末端(PE150)测序(Illumina,圣地亚哥,加利福尼亚,美国)。这个 Hi-C 文库生成了大约 64.61 Gb(约 115 倍深度)的清洁数据。
基因组survey分析
使用 GenomeScope v1 生成了大约 31.47 Gb 的清洁读数用于基因组调查,参数为“k = 15, 17, 19, 21, 23; length = 100; and max coverage = 10000”。我们得到了一个估计的基因组大小为 470.69 Mb 至 513.17 Mb,异质性为 0.29% 至 0.41%。
二倍体解析染色体规模基因组的去 novo 组装
在去除接头后,使用 hifiasm v0.16.1(默认参数)利用 HiFi 读数组装初级拼接体数据集。其次,使用 minimap2 v2.24 将所有 CCS 读数重新映射到上述组装的拼接体上,参数设置为 -secondary = n -cx map-pb。第三,使用 Longshot(默认设置)调用变异。第四,使用 WhatsHap(默认参数)实现变异分相。第五,根据分相的变异将 CCS 读数分为两个二倍体(hap1 和 hap2)以及一个未分配组。第六,将 hap1 和 hap2 的未分配组的 CCS 读数分别独立使用 hifiasm v0.16.1(默认参数)进行组装。为了基因组支架,使用 BWA v0.7.10 将干净的 Hi-C 数据映射到 hap1 和 hap2 的拼接体上。使用 HiCExplorer 计算映射读数和交互对。对于 hap1,共有 120.89 百万读数被唯一映射,其中 51.16 百万为有效交互,占比 42.32%。对于 hap2,有 119.03 百万读数被唯一映射,50.58 百万读数与其他配对进行了交互,有效数据占比 42.49%。其次,使用有效交互读数配对通过 HiCAssembler(选项:-min_scaffold_length 100000; -bin_size 20000; -misassembly_zscore_threshold 1.0; -num_iterations 4; -num_processors 30)将组装的二倍体解析拼接体聚类和定向到拟染色体上。使用 Juicebox 工具(GitHub - aidenlab/Juicebox: Visualization and analysis software for Hi-C data -)手动微调拟染色体。这一最终组装结果产生了总共 562.99 Mb(拼接体 N50 = 34.10 Mb)分配到 16 个拟染色体的 hap1,其中 527.20 Mb 能被定向,占二倍体基因组大小的 93.64%,而 hap2 的 526.40 Mb 被定向,占组装序列的 561.07 Mb(拼接体 N50 = 33.91 Mb)的 93.82%(表 2;图 2c,d)。热图显示了 20 kb 分辨率的基因组读数配对的交互作用(图 2a,b)。
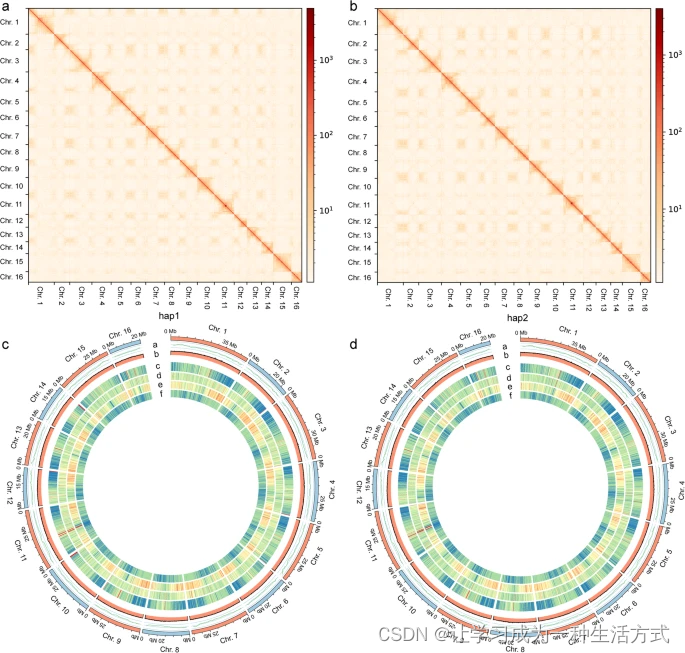
Hi-C 热图和 Circos 图表用于 hap1 和 hap2。(a,b)显示了以 20 kb 为单位的 Hi-C 数据的交互链接。交互强度使用从浅黄色到深红色的颜色表示,这些颜色代表从低到高的 Hi-C 交互频率。(c,d)Circos 图显示了 hap1 和 hap2 基因组,窗口大小为 500 kb。这个 Circos 图包括六个组成部分:(a)LAI(LTR 组装指数)值;(b)GC 含量密度;(c)基因密度;(d)Copia 类反转座子密度;(e)Gypsy 类反转座子密度;和(f)TEs(可移动元素)的密度。
基因组范围内重复序列的注释
采用了一个综合的转座子(TE)预测工具,Extensive Denovo TE Annotator(选项:-t 20 -step all -sensitive 1 -anno 1),用以构建一个非冗余TE库,并进行Helitron和长末端重复(LTR)的识别。使用 TIR-Learner v2.5 在默认推荐下搜索末端倒置重复(TIR)。使用 RepeatMasker v4.0(参数:-nolow -no_is -norna -engine wublast)预测基因组的最终重复序列。最终,我们在hap1和hap2组装中分别识别出46.26%和46.23%的重复元素,包括LTR类型(4%-8%)、TIR类型(0-5%)、非LTR类型(约0.50%)和非TIR类型(2%-9%)(表3;图2c,d)。
基因模型预测和功能注释
蛋白编码基因模型预测结合了三个步骤。首先,使用 AUGUSTUS v3.4 和 GeneMark-ES v4.68 通过自发预测搜索基因模型。其次,使用 GeMoMa v1.3 进行同源预测。然后,使用 PASA(参数:-align_tools gmap, -maxIntronLen 20000)通过 RNA-seq 进行基因预测。最终,为hap1注释了41039个基因模型,为hap2注释了39744个基因模型(表4;图2c,d)。对于功能注释,将上述基因模型序列翻译成蛋白序列,并随后与 COG v2.1、GO、Pfam 和 KEGG 数据库进行比较。总共有34617个hap1和34562个hap2的非冗余基因模型通过这些生物数据库进行了注释,分别占所有基因模型的84.35%和86.96%(表4)。hap1和hap2的基因模型数量均高于 Chandler(32498)、Serr(Payne × PI 159568)(31425)和 Ding 等人(33430)报告的数量,低于 Chandler(37554)和 Zhongmucha-1(39432)报告的数量。
非编码 RNA 注释
tRNAscan-SE v2.0(默认设置)、barrnap v0.9(选项:-kingdom euk -threads 8)和 Infernal v1.0(选项:-Z $Z -cut_ga -rfam –nohmmonly -fmt 2)结合 Rfam 分别用于 tRNA、rRNA 和 miRNA/snoRNA 的搜索。总的来说,分别在hap1和hap2的基因组中发现了1817个(500个tRNA,72个rRNA,207个miRNA和1038个snoRNA)和1919个(500个tRNA,104个rRNA,210个miRNA和1105个snoRNA)非编码RNAs(表4)。
简单序列重复(SSR)识别和变异调用
使用 TBtools v 1.09875 在默认设置下分析 hap1 和 hap2 以识别 SSR。我们为hap1识别了358869个SSR,为hap2识别了358104个SSR。此外,我们使用 hap2 作为查询,将其使用 minimap2 v2.24 映射到 hap1 上(设置为 -eqx -ax asm5),然后使用 SyRI 在默认设置下进行变异检测。随后,我们检测到了21个倒位,40个易位,575个存在/不存在变异(PAVs),在hap1中2个复制,在hap2中60个复制(表5;图3)。值得注意的是,PAVs的长度在hap1中为274,629 bp,在hap2中为274,840 bp,分别占hap1和hap2中303和272个基因(参见存储库文件)。类似地,hap1与三个已发布的 Juglans 基因组之间的结构变异也被识别(参见存储库文件)。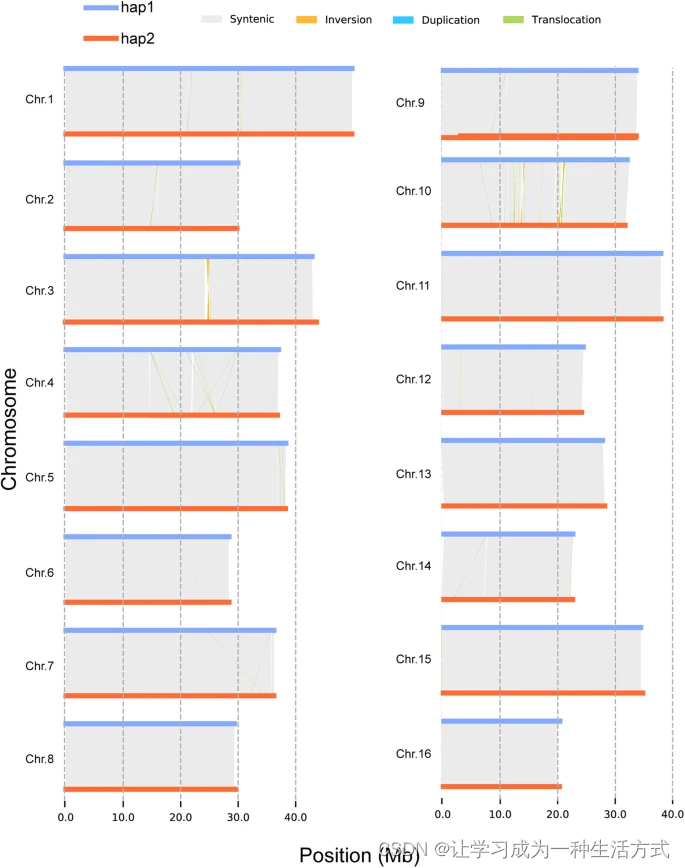
二倍体间的结构变异(SV)分布。水平的蓝线表示 hap1 的染色体,红线表示 hap2 的染色体。灰色、橙色、浅蓝色和绿色线分别表示同源区域、倒置区域、重复区域和易位区域。
冷胁迫下的等位基因特异性表达基因(ASEGs)
为探索ASEGs对冷胁迫的响应,从野生胡桃树种仁衍生的三叶期幼苗在4°C的气候室中(上海博讯工业有限公司,上海,中国)进行处理。处理后0小时、3小时、6小时、12小时和24小时分别采集约0.5克叶片,进行三次生物重复,立即放入液氮中快速冻存。构建了RNA-seq文库,并进行了测序(见方法:植物材料准备、Illumina短读序列库构建和测序)。使用MCScanX识别hap1和hap2之间的共线块基因对(默认设置)。移除涉及支架的共线基因对。随后,使用BLAST检查共线基因对,相似度在90%至100%之间。最终我们获得了18770个基因对。使用HISAT2 v2.20将清洁的RNA-seq数据映射到胡桃基因组,并使用StringTie v2.1.2进行表达量化,TPM值用于表达量化。使用DESeq 2根据标准|log2Fold Change| > 1和调整后的p < 0.05来确定ASEGs。我们分别在0小时、3小时、6小时、12小时和24小时获得了358、457、465、494和386个ASEGs,占所有识别到的共线基因的约2%(见存储库文件;图4)。在所有ASEGs中,234个是共有的,分别在0小时、3小时、6小时、12小时和24小时的样本中特异性检测到12、37、22、32和20个(图5b)。我们还注意到,更频繁的ASEGs主要在hap2上表达(图5a),这表明在野生胡桃的冷胁迫下,ASEGs的等位基因表达模式不协调。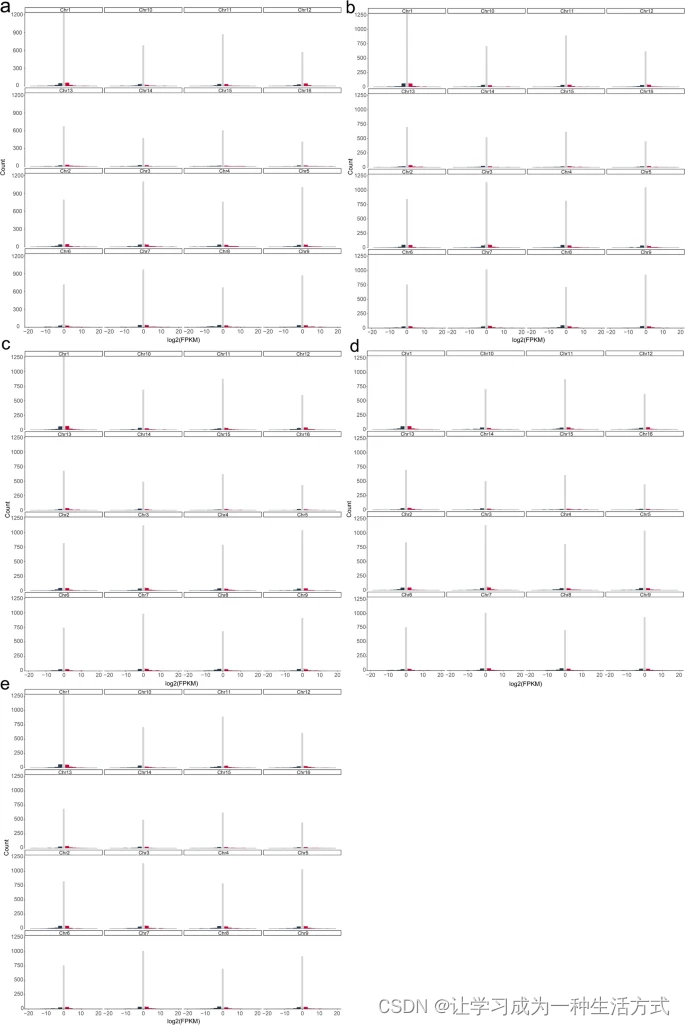
在0小时(a)、3小时(b)、6小时(c)、12小时(d)和24小时(e)的同源染色体对中,发散等位基因的双等位基因表达的直方图。黑色柱表示hap1占优势。红色柱表示hap2占优势。灰色柱表示表达均等。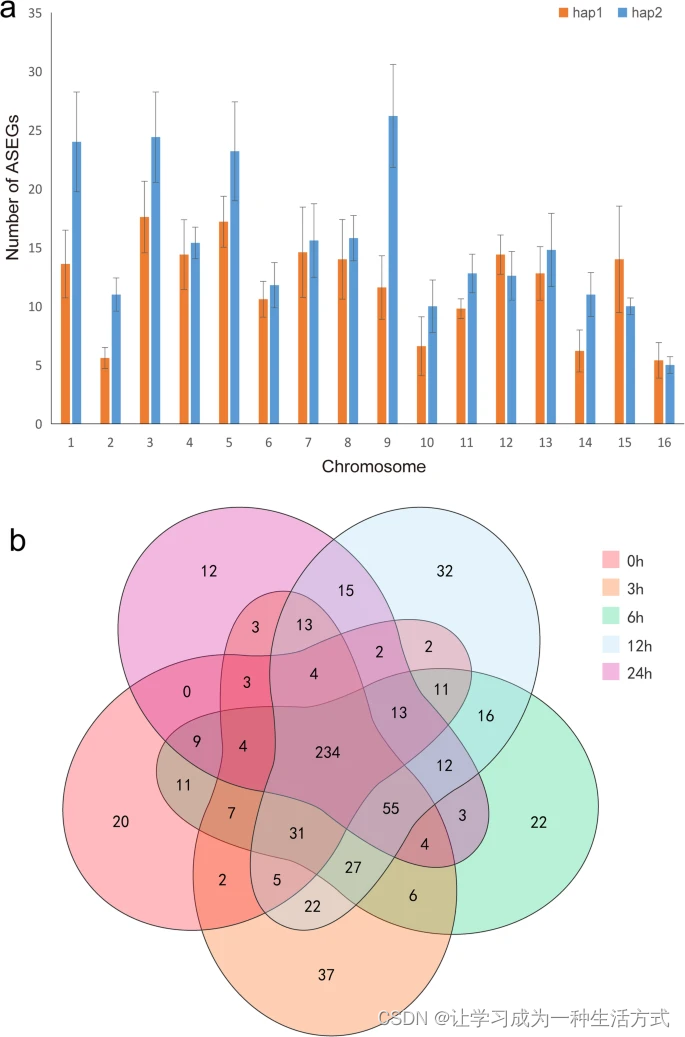
冷胁迫下hap1和hap2中的等位基因特异性表达。(a)在冷胁迫下五个时间点(0小时、3小时、6小时、12小时、24小时)的二倍体染色体上的等位基因特异性表达基因(ASEGs)。ASEGs的数量显示为平均值 ± 标准差。(b)冷胁迫下五个时间点的ASEGs的维恩图。数字表示不同时间点的独特和共有的ASEGs数量。
数据记录
用于基因组survey的Illumina读取数据、RNA-seq读取数据和Hi-C读取数据已存储于序列读取档案库(SRA)(登录号分别为:PRJNA858167、PRJNA859241和PRJNA858917)【84,85,86】。HiFi长读取数据已在国家基因组数据中心(Home - National Genomics Data Center)和SRA中共同存储,登录号分别为CRA007543和PRJNA947329【87】。野生胡桃(Juglans regia L.)的基因组组装已在Figshare平台(haplotype genome sequence)【88】和GenBank(登录号为GCA_034508915.1和GCA_034509015.1)存储【89,90】。冷胁迫样本的RNA-seq读取数据已存储在SRA中,登录号为PRJNA942426【91】。
技术验证
测序读取质量控制
对于基于Illumina的测序数据(用于基因组调查、Hi-C读取和RNAseq读取),按照以下步骤处理原始读取数据以获取清洁数据:(1)过滤接头;(2)移除含有超过10%未识别核苷酸的读取;(3)移除含有超过50% Q值≤10的碱基的读取。然后,根据插入大小、GC含量、Q30值、质量分布和碱基组成评估清洁数据。特别是,我们使用HiCUP【92】(v0.6.1)进行预处理和评估Hi-C文库质量。在调用HiCUP_truncater脚本修剪HindIII的限制酶位点后,Hi-C清洁数据被对准到二倍体基因组,只有映射得分至少为20的唯一映射序列被用于后续处理。对于HiFi长读取,同一模板链的子读取被一致地校正,过滤出适配器和低质量读取。在处理前,随机选取的2000个长读取被用于通过BLAST【80】对Nt数据库(ftp://ftp.ncbi.nih.gov/blast/db)进行污染评估,阈值为P < 1e-05。
二倍体基因组组装评估
BUSCO【45】v5的评估显示,hap1和hap2中成功组装的Embryophyta基因超过94%(图6),表明这些二倍体解析基因组的完整性符合已发布基因组的标准【29】。hap1和hap2的LAI【44】值分别为13.36和13.41。hap1和hap2的CC比例【93】分别为43.25和42.94。此外,我们使用hap1和hap2作为查询对象,对照两个已发布的基因组【29,71】使用minimap2 v2.24进行映射,设置为-eqx -ax asm5。我们的二倍体基因组与这两个已发布的基因组显示出良好的共线性【29,71】(图7)。最后,使用修改后的calc_switchErr流水线【32,94】通过开关错误的百分比评估二倍体解析组装的分相准确性。由于没有可用的10x Genomics连接读取,从分相评估流水线中排除了标准一致性分相SNP步骤。共检测到184957个SNP,其中33061个为开关SNP,表明开关错误率为17.87%,与马铃薯(Solanum tuberosum L.)基因组的17.1%相似,低于苹果(Malus domestica cv.Gala)的22.2%和香草(Vanilla planifolia)的44.0%【94】。我们进一步使用已发布的Juglans regia L.品种Chandler的Nanopore测序数据(SRR10001245)【28】评估分相准确性。首先使用野生胡桃的基因组重测序数据,使用Ratatosk【95】v0.7.0进行ONT数据的校正;然后,使用同样的方法计算开关错误。最终,我们得到了6.25%的开关错误。总体来说,这些结果表明我们获得了一份参考级高质量的野生胡桃二倍体解析基因组。
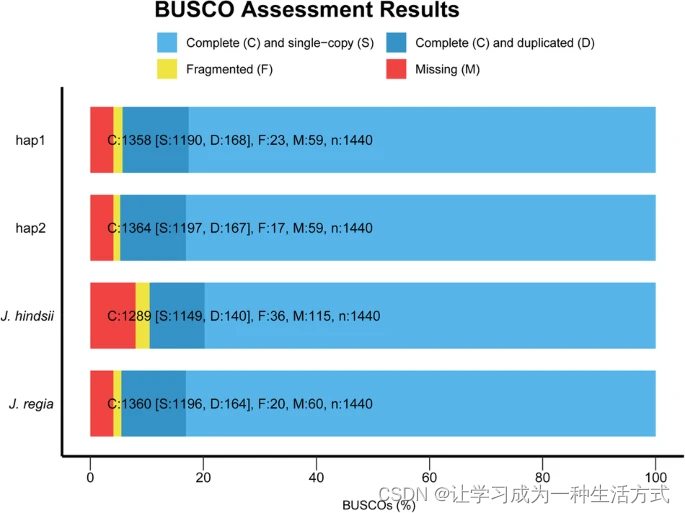
BUSCO评估的总结评估条形图,用于hap1、hap2和已发布的Juglans基因组(Juglans Genome - Genome Projects)。浅蓝色表示完整且单拷贝基因的百分比,而较深蓝色表示完整且重复基因的百分比;黄色表示片段化基因的百分比;红色表示组装中缺失基因的百分比。
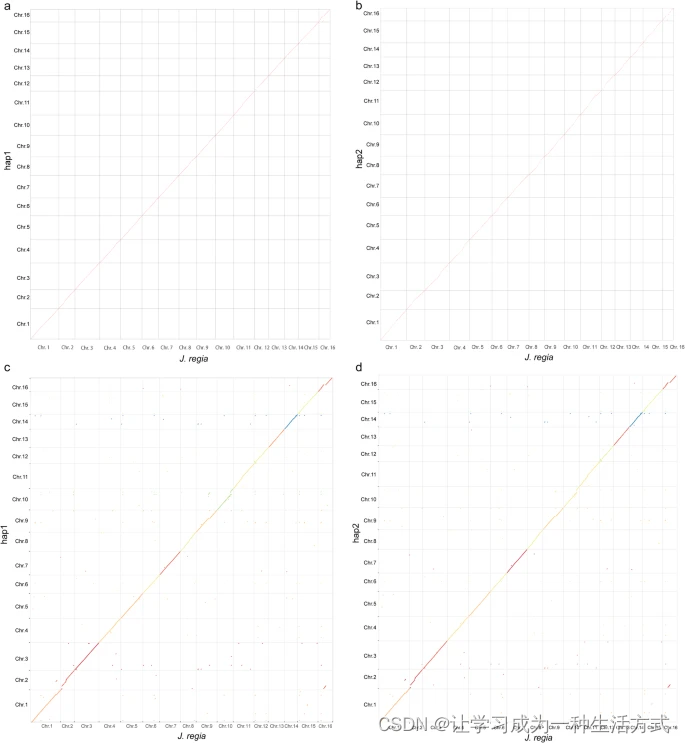
二倍体与已发布的Juglans regia L.基因组之间的共线性评估29,71。(a)hap1与已发布的Juglans regia L.基因组29之间的共线性。(b)hap2与已发布的Juglans regia L.基因组29之间的共线性。(c)hap1与已发布的Juglans regia L.基因组71之间的共线性。(d)hap2与已发布的Juglans regia L.基因组71之间的共线性。为了区分这两个已发布的基因组,使用不同颜色标记了染色体71的共线性。
已发布的QTL验证
为了进一步验证基因组的准确性,我们下载了已发布的Juglans regia L.的简化代表性测序数据,这些数据用于高密度遗传图谱构建和QTL定位【96】。总共有2540个标记(共2577个,占98.56%)通过特定长度扩增片段测序(SLAF-seq)开发,在遗传图谱上可以映射到hap1的基因组,相似度>90%且E阈值<E-10(见存储库文件【78】)。特别地,作者识别的一个抗炭疽病的QTL含有10个标记,被发现最佳映射到hap1的chr1:14148922–14220999区域,该区域大约721 kb(表6)。对hap2也做了类似的发现。这些结果表明了这种野生胡桃亚基因组的高准确性。

















 1089
1089

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









