










The gap-free genome of mulberry elucidates the architecture and evolution of polycentric chromosomes
桑树无间隙基因组阐明多中心染色体的结构和进化,就是一个染色体多个着丝粒~

摘要
桑树是全球蚕桑产业的基本组成部分,其对我们的健康和环境的积极影响不可低估。然而,之前报道的桑树参考基因组存在未组装或未定位的序列。在这里,我们报道了桑树种植川桑的端到端无间隙参考基因组的组装和分析,该基因组已成为桑树基因功能研究和遗传改良的重要参考。本研究产生的桑树无间隙参考基因组为我们研究着丝粒的结构和功能提供了前所未有的机会。我们的结果显示,所有桑树的着丝粒区域都共享具有不同拷贝数的保守的着丝粒卫星重复序列。引人注目的是,我们发现川桑是一种具有多中心染色体的物种,也是迄今为止唯一报道的多中心染色体物种。我们提出了一个解释新着丝粒形成机制的有力模型,并解答了桑树种染色体融合-裂解周期的未解之谜。我们的研究为桑树种的功能基因组学、染色体进化和遗传改良提供了新的见解。
引言
桑树不仅在蚕桑产业中占据重要地位,而且在环境保护中也扮演着关键角色。桑树的首个草稿基因组——川桑,于2013年报道[1]。在过去十年中,川桑已成为桑树基因[2]、表型[3]、基因组学[4, 5]及其他组学研究[6]的重要模型系统。尽管近年来报告了几个高质量的桑树基因组,包括川桑[5, 7]、白桑[8]、印度桑[9]和云南桑[7],但仍有许多未组装或未定位的序列。
作为基因组项目的最后边疆,着丝粒区域是最后也是最难处理的部分。它们包含极高含量的重复序列,这阻碍了其组装。在过去两年中,包括水稻[10–12]、拟南芥[13–15]、香蕉[16]、西瓜[17]、大麦[18]、苦瓜[19]、甘蓝[20]、红背桃[21]和草莓[22]在内的一些植物种的端到端(T2T)或无缝基因组已被报道。令人惊讶的是,这些物种的所有着丝粒区域都被成功组装。考虑到桑树具有多种不同的倍性水平[23],并且着丝粒不仅在细胞分裂中发挥作用,还决定了大规模基因组结构和染色质组成[24],研究完整的着丝粒可以加深我们对这一物种进化过程的理解。然而,缺乏桑树的T2T参考基因组阻碍了对其有趣的着丝粒组织和染色体进化的详细研究。
在本研究中,通过结合PacBio HiFi的高覆盖率和精确的长读序列数据以及Oxford Ultra-long测序,组装了桑树的首个T2T无间隙参考基因组。引人注目的是,我们报告了一个多中心基因组的完整特征,这是迄今为止唯一报道的多中心基因组。我们进一步提出了一个适用于川桑染色体融合-裂解周期的模型。我们的结果揭示了这一物种的基因组组织和染色体进化,为桑树的进一步遗传研究和遗传改良提供了科学参考。
结果
川桑的端到端基因组
Mnot-SWU估计的基因组大小为389.35 Mb(表S1,请参阅在线补充材料)。为了开发川桑的高质量基因组组装,我们使用了通过各种测序平台生成的各种数据。总共产生了15.06 Gb(约40×覆盖率)的HiFi读数,这些读数是由PacBio Sequel II平台生成的(表S2,请参阅在线补充材料),以及由ONT平台生产的43.10 Gb(约110×覆盖率)的ONT超长读数(表S3,请参阅在线补充材料)。HiFi和ONT超长读数的N50长度分别大于15.75 kb和100.71 kb(表S2和S3,请参阅在线补充材料)。通过HiFi和ONT超长读数进行初步组装,生成了四组初级基因组连锁体(表S4,请参阅在线补充材料)。Hifiasm [25] 生成的组装基因组不仅N50、N90和最大连锁体长度是其他组装版本的2-4倍,而且含有最少的连锁体数量。因此,由Hifiasm生成的组装基因组被设定为主干基因组。同时,我们生成了40.69 Gb(约100×覆盖率)的HiC测序数据,用于将连锁体锚定到染色体上(表S5,请参阅在线补充材料)。之后,所有连锁体都能锚定到六条染色体上,我们生成了一个新版本的组装基因组,包括32个连锁体,其中包括26个间隙(表S6,请参阅在线补充材料)。使用ONT读数的其他基因组组装版本来填补主干基因组的间隙。填补所有26个间隙后,我们生成了一个无间隙的参考基因组,名为Mnot-SWU,包含六条染色体,总长度为410.45 Mb,N50长度达到75.38 Mb(表1,图1A;图S1和S2,请参阅在线补充材料)。与先前报道的基因组相比,川桑的基因组质量有了显著提高(表1;表S7,请参阅在线补充材料)。新的组装还纠正了先前报道的基因组中的一些组装错误,包括定向错误、组装错误和不明确区域(图1A)。最后,使用保守的端粒重复序列(5’-CCCTAAA/TTTAGGG-3′)作为序列查询,鉴定出十个端粒(图1A;表S8,请参阅在线补充材料)。
Characteristics of the *Morus notabilis* genome, Mnot-SWU.
| Genomic feature | Mnot-SWU |
|---|---|
| The total size of assembled contigs (Mb) | 410.45 |
| Number of contigs | 6 |
| Contig N50 (Mb) | 75.38 |
| Gaps | 0 |
| Number of telomeres | 10 |
| BUSCOs (%) | 98.51 |
| Number of genes | 27 413 |
| LTR assembly index score | 19.26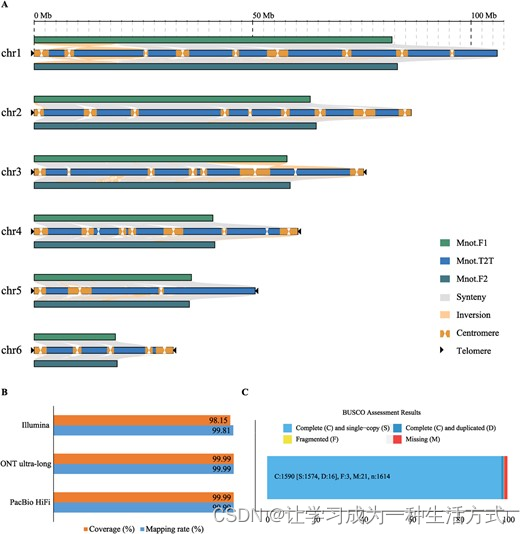 |
无间隙参考基因组桑树Mnot-SWU。A 对Mnot-SWU、Mnot.F1和Mnot.F2进行同源性分析。B 不同测序平台读数的映射率和覆盖率。C 使用胚植物_odb10数据库对基因组组装和基因模型预测的完整性进行BUSCO评估。
Mnot-SWU基因组的质量通过多种方式进行评估。首先,将来自三种不同测序平台(如PacBio HiFi、ONT超长和Illumina)的所有读数映射到Mnot-SWU基因组上,所有映射率均达到99%,表明组装的基因组高度连续且精确度高(图1B;表S9,请参阅在线补充材料)。通过多种方法预测了27413个基因模型,其中98.51%的1614个参考基因集可以在27413个基因模型集中被识别(图1C;表S10,请参阅在线补充材料)。在注释这些编码基因时,我们发现共有94.90%的基因能与公共数据库对齐,包括NR、Swiss-Prot、KEGG、InterPro、Pfam和GO(表S11,请参阅在线补充材料)。Mnot-SWU的LTR组装指数(LAI)为19.26(表1)。我们还评估了基于k-mer的Mnot-SWU基因组质量估计,基因组的质量值为40.24。就所有染色体而言,质量值范围在39.18到41.72之间(表S12,请参阅在线补充材料)。综合这些结果,此处呈现的端到端Mnot-SWU基因组具有最高的可靠性和质量。
端到端Mnot-SWU基因组提供了前所未有的机会,可以识别所有转座元素、其他重复序列和非编码基因。因此,共识别出了242.97 Mb的散布重复序列(表S13,请参阅在线补充材料),占组装基因组的59.20%。
Mnot-SWU的每条染色体被分割成若干富含串联重复序列的区间
基于Mnot-SWU与另外两个先前报道的基因组Mnot.F1 [5]和Mnot.F2 [7]之间的同源性结果,每条染色体上均存在许多未对齐的区域,这些是在Mnot-SWU中新识别的(图1A)。为了清楚地展示它们,我们重新绘制了Mnot-SWU与Mnot.F1基因组之间最长染色体(chr1,106.00 Mb)的同源性(图2A)。结果中可以清晰地看到九个空白区域。通过对基因组之间的同源性分析,发现其他五条染色体也存在相同现象,空白区域的数量从三到九不等(图S3,请参阅在线补充材料)。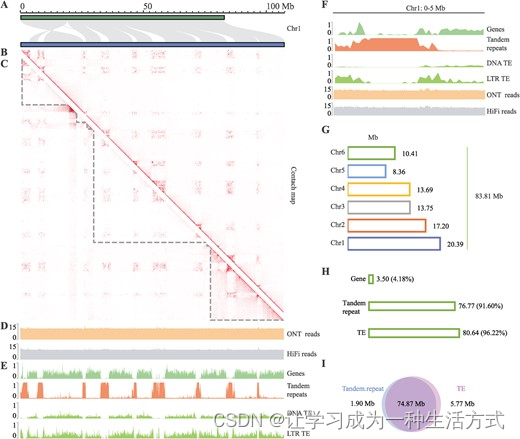
Mnot-SWU染色体被分割成几个富含串联重复的区域。A 在Mnot-SWU(蓝色)和Mnot.vF1(绿色)之间chr1的详细同源性视图。灰线表示两个基因组之间chr1的同源区域。空白区域代表在Mnot-SWU基因组中新组装的区域,这些区域在之前的基因组中不存在。B 基于Mnot-SWU中HiC数据的chr1染色体互作热图。C 基于初步组装中HiC数据的chr1染色体互作热图。每个带有灰色虚线的三角区域代表初步组装中chr1的一个连锁体。D chr1中ONT超长(上)和PacBio HiFi(下)读数的覆盖率。应用了对数变换(log2),y轴给出了变换后的值。E chr1详细视图显示了聚集的基因组特征分布。基因组特征分布的窗口大小设置为100 kb。从上到下依次是基因、串联重复、DNA型TE和LTR型TE。F chr1的放大视图显示了一个5 Mb的区域,靠近初步组装chr1的第一个连锁体的边缘。所有值都是基于100 kb的窗口计算的。G Mnot-SWU基因组中每条染色体新组装区域的大小。H Mnot-SWU基因组中新组装区域的基因组特征内容。I 新组装区域中串联重复和TE的比较。
当我们进行染色质互作分析时,出现了一个令人惊讶的现象。一般来说,单中心物种中的典型染色质互作在常染色质和异染色质中表现为大规模的区室化和某种程度的端粒到着丝粒轴线[26]。我们也在所有染色体中观察到了大规模的区室,但其数量与典型的单中心物种不同(图2B;图S4,请参阅在线补充材料)。例如,我们还在chr1中发现了九个大规模的区室和另一个聚集轴线(图2B)。chr1中染色质互作的分布模式立即引起了我们的注意,因为这种模式与Mnot-SWU和Mnot.F1之间chr1的先前同源性分析结果一致(图2A和B)。其他五条染色体也显示出同样的特征(图S3和S4,请参阅在线补充材料)。
分析了初步组装基因组中chr1的染色质互作,这一现象得到了证实。如图2C所示,初步组装的chr1包含七个连锁体,Mnot-SWU基因组中的大部分新组装区域(图2A中的空白区域和图2B和C中的聚集轴线)位于其中一个连锁体内。HiFi读数和ONT超长读数在这些新组装的区间内均匀分布(图2D),这也强有力地证明了每个新组装区域的连续性和高质量。这些结果在其他染色体中也是非例外的(图S5和S6,请参阅在线补充材料)。
值得注意的是,每条染色体上的基因组特征分布也显示了相似的模式。首先,川桑的基因集中在九个区间中(图2E;图S7,请参阅在线补充材料)。这些串联重复大多在Mnot-SWU基因组的新组装区域中被识别,这些区间中大部分100 kb的箱中串联重复的覆盖率达到了100%(图2E;图S8,请参阅在线补充材料)。我们还计算了DNA和LTR转座元素在100 kb窗口中的覆盖率。两者的模式与基因的模式相似(图2E;图S9,请参阅在线补充材料)。所有基因以及DNA和LTR转座元素都被串联重复分割成九个区间(图2E;图S8,请参阅在线补充材料)。chr1的放大视图显示了一个0-5 Mb的区域,位于初步组装chr1的第一个连锁体的边缘。一个连锁体边缘的基因组特征分布模式与同一连锁体内部或其他连锁体的分布模式没有显著差异(图2F)。
这些基因组特征的分布模式和新组装区域在所有六条染色体上的情况促使我们进一步分析这些区间特征。我们计算了所有染色体上所有新组装区域的大小,Mnot-SWU基因组中共新组装了83.81 Mb的区间(图2G)。从chr1到chr6,新组装区间的大小分别为20.39、17.20、13.75、13.69、8.36和10.41 Mb。除chr5外,每条染色体上新组装区间的大小与相应染色体大小正相关。随后,我们重点研究了所有六条染色体上这些新组装区域的组成(图2H)。共3.50 Mb(83.81 Mb中的4.18%)的新组装序列被注释为与基因相关的序列,包括UTR、外显子和内含子。关于其他序列,76.77 Mb(91.60%)的新组装序列可以归类为串联重复,总共80.64 Mb(96.22%)的这些序列被视为TEs。我们计算了串联重复和TEs的重叠,经过比较串联重复和TEs的位置,我们发现74.87 Mb的序列,占76.77 Mb串联重复中的97.53%,同时被定义为串联重复和TEs(图2I)。
基于上述结果,川桑的每条染色体被划分为几个富含串联重复的区间。
川桑是一种基于重复序列的多中心染色体物种
川桑的端到端基因组为我们研究其着丝粒提供了绝佳的机会,并允许我们在Mnot-SWU基因组中解析完整的着丝粒重复序列阵列。首先,我们分析了川桑的着丝粒序列组织。连续且最丰富的重复单元被视为每条六条染色体中的着丝粒串联重复。结果表明,我们在每条染色体上鉴定了六个着丝粒串联重复,每个长度为82 bp(表S14,请参阅在线补充材料)。由于这六个着丝粒串联重复的全球同一性范围为93%至100%(图3A),这些重复被命名为m3cp并用于进一步分析。我们计算了每条染色体的着丝粒m3cp重复的总量(图3B),与相应染色体大小正相关;其值分别为chr1的11.96 Mb、chr2的10.60 Mb、chr3的9.74 Mb、chr4的7.64 Mb和chr6的6.43 Mb。只有2.86 Mb的m3cp阵列在chr5中被注释。这些重复在新组装区域中的比例也进行了评估(图3C)。在Mnot-SWU基因组的chr1、chr2、chr3、chr4和chr6中,分别有96.91%、90.60%、89.43%、93.12%和96.30%的m3cp重复是新鉴定的。在Mnot-SWU基因组的chr5中,新鉴定的m3cp重复的比例仅为74.87%。
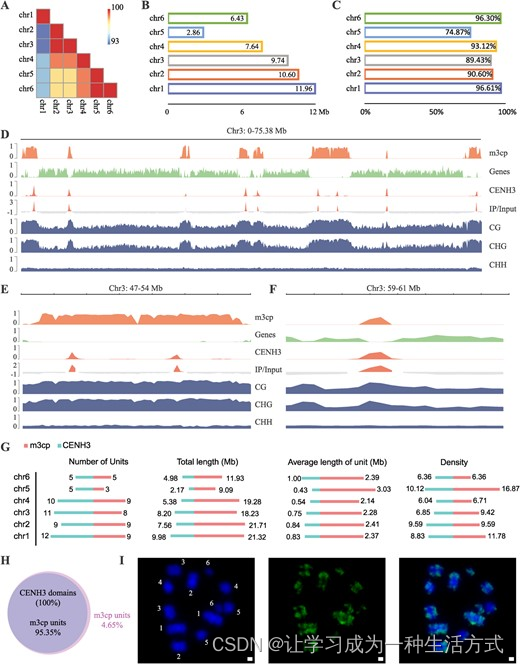
Morus notabilis中基于重复序列的多中心染色体的特征和遗传组成 A 六条染色体中m3cp序列同一性的热图。B 每条染色体的m3cp重复总量。C m3cp重复位于新组装区域的比例。D chr3的全局视图,展示了m3cp阵列、基因、CENH3区域、IP与输入的比率以及三种5mC甲基化类型(CG、CHG和CHH)的分布。所有值均基于100 kb的窗口计算。E chr3的特写视图,展示了包含两个着丝粒单元的7 Mb区域,这些单元由两个CENH3区域和m3cp阵列组成。F chr3的放大视图,展示了一个2 Mb区域,其中CENH3区域长度大于m3cp阵列。G 川桑中m3cp和CENH3区域的特征。从左到右,单位数量、总长度、单位的平均长度、每条染色体上m3cp阵列和CENH3区域的密度。H m3cp阵列和CENH3区域的间隔比较。I 通过FISH确定m3cp的染色体位置。从左到右,DAPI,FITC和合并图像。
Morus notabilis是一种多中心染色体物种。我们重新分析了川桑的染色质结构,发现chr3的基因沿整个染色体集中在几个区域(图3D)。相反,所有的着丝粒m3cp重复都位于基因密度低的区间中。显然,沿着chr3染色体有八个富含着丝粒m3cp重复的区间。其他五条染色体中的着丝粒m3cp重复富集区间的数量范围为三到九(图S10和表S15,请参阅在线补充材料)。同时,我们还进行了ChIP-seq实验以确定川桑染色体中着丝粒组蛋白H3(CENH3)的结合区域(图3D;图S10,请参阅在线补充材料)。如图3D所示,我们在chr3染色体上检测到了九个聚集的CENH3结合区域。在chr3的47-54 Mb区域中检测到了两个CENH3区域(图3E)。在所有六条染色体中,大多数m3cp阵列的长度都大于CENH3区域的长度(图3D)。图3F显示了其他m3cp阵列的放大视图,其长度大于CENH3区域。注意整个染色体的5mC甲基化状态(图3D,E和F)。显著的是,在CG和CHG背景下,m3cp富集区域的甲基化比其他区域更为明显。相反,在着丝粒重复区域中,mCHH的模式是均匀的。
通过多种方式比较了着丝粒m3cp重复和CENH3区域的特征(图3G)。每条染色体上m3cp和CENH3区域的总单位数量分别为五到九和五到十二。关于这些单位的密度,它们的模式与单位数量相似。总体而言,每条染色体上的m3cp阵列总长度(9.09–21.32 Mb)大于CENH3区域的总长度(2.17–9.98 Mb)。单位平均长度也显示出相似的现象,m3cp阵列为2.14–3.03 Mb,CENH3区域为0.43–1.00 Mb。m3cp阵列的总数为43,比CENH3区域的总数52少。值得注意的是,100%的CENH3区域可以映射到93.55%的m3cp阵列区域(图3H)。我们还对这两类间隔进行了Fisher检验,得到的P值低于0.05;这表明在全基因组范围内,m3cp重复与CENH3区域之间存在显著关联。换句话说,ChIP-seq结果也证明了m3cp重复是着丝粒串联重复和川桑中主要的CENH3结合位点。我们进一步进行了荧光原位杂交(FISH)以展示m3cp重复在所有六条染色体中的分布(图3I)。结果表明,在所有六条染色体中都有多个m3cp重复信号,表明m3cp重复在每条染色体中分布在多个位置。这使我们推断出川桑是一种具有多中心染色体的物种。
川桑中特定chr5的特征和遗传组成
chr5是川桑的一条特定染色体。如上所述,在chr5上仅鉴定出了总计9.09 Mb的m3cp重复阵列,而其他五条染色体的值在11.93至21.71 Mb之间(图3G)。考虑到chr5的大小是chr6的1.6倍,m3cp重复的总量仅为chr6的76.19%。此外,在chr5上仅鉴定出三个着丝粒(图4A;表S15,请参阅在线补充材料),而其他五条染色体上的着丝粒数量在五到九之间。m3cp重复阵列和CENH3区域的单位平均长度和密度与其他五条染色体相比有显著差异(图3G)。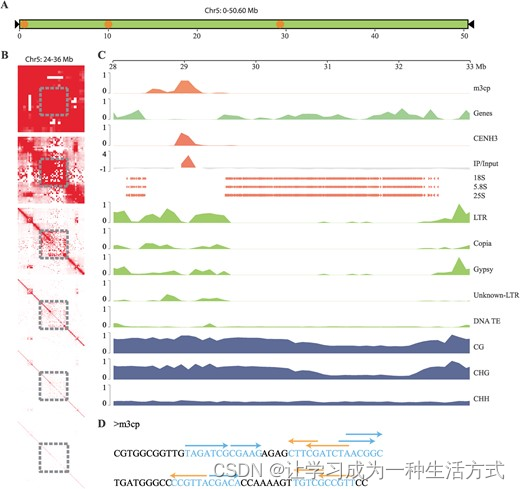
Morus notabilis 特定染色体 5 的特征和遗传组成 A 染色体模型图的chr5。黑色三角形表示端粒,橙色圆圈表示着丝粒的大致位置。B 已组装的24-36 Mb的chr5染色体的接触图。灰色方块表示chr5的28-32 Mb区域。C chr5中28-32 Mb区域的基因组特征分布全局视图。所有值均基于100 kb的窗口计算。D m3cp重复序列中的二聚对称性模式。蓝色和橙色箭头分别表示正向或反向方向。
在chr5染色体中鉴定出了一个异常的染色质互作区域。我们基于不同分辨率水平下的HiC数据(包括500、250、100、50、25和10 kb)对这条染色体进行了染色质互作分析。图4B显示了chr5的24-36 Mb的接触图。当分辨率设为250 kb时,出现了一个染色质互作较少的区域。随着分辨率从250 kb降低到10 kb,这个区域向两侧扩展。我们计算了整个chr5中基因组特征的覆盖率,基于结果,与其他区域相比,这一区域内丰富的LTR转座子元素(图S11,请参阅在线补充材料)。在这一区域还发现了带有CENH3区域的着丝粒。此外,本应分布在一个簇中的rRNA基因被这一着丝粒分成了两个簇(图4C)。
我们重新分析了着丝粒m3cp重复序列的特征;图4D显示了着丝粒m3cp重复序列中的DNA二聚对称性模式。值得注意的是,为了简化,图中仅显示了长度大于5 bp的二聚对称性。当二聚对称性长度设定为大于3 bp时,95.12%的m3cp重复长度展示了二聚对称结构(表S16,请参阅在线补充材料)。
讨论
在此,我们报告了一种无间隙的端到端高质量桑树参考基因组。这一新组装的基因组不仅对染色体进化研究有力,也有助于桑树的遗传改良。
川桑的T2T基因组为研究桑树基因组提供了前所未有的机会
桑树的第一个草稿基因组于2013年报道[1]。尽管近年来也报告了一些高质量的桑树参考基因组,但它们仍然未组装或未定位[5, 7]。基因组组装可以比作一个有许多碎片的拼图。要组装一个完美的基因组,我们需要更加关注测序的准确性和连续性。最近,使用多种测序策略成功组装了接近完整或无间隙的植物基因组[27, 28]。在本研究中,我们使用包括PacBio HiFi和Oxford Ultra-long测序等新型和尖端测序技术,首次实现了桑树的T2T参考基因组(Mnot-SWU)。结果表明,与之前报道的桑树基因组相比,在所有六条染色体上新组装了总计83.81 Mb的序列。此外,植物中的端粒通常由相对保守的微卫星序列串联重复组成[29]。然而,在Mnot-SWU基因组中,未在chr1和chr2的3′端检测到保守的端粒重复序列。在水稻[11, 12]、葡萄藤[30]、苦瓜[19]和杜鹃花[31]的T2T组装的某些染色体中也发现了类似的现象。考虑到在其他植物中已鉴定出其他类型的端粒重复序列,如达尔马西亚风信子[32]和大戟属植物[33],因此我们假设桑树中可能也存在其他类型的端粒重复序列。Mnot-SWU基因组为研究整个桑树基因组提供了前所未有的资源。
迄今为止,川桑是唯一报道的多中心染色体物种
这里产生的T2T参考基因组促进了首次在植物中对着丝粒进行全球准确评估,因为着丝粒始终非常难以组装。最近,一些物种也组装了接近完整基因组的着丝粒,包括水稻[10-12]、拟南芥[13-15]、香蕉[16]、西瓜[17]、大麦[18]、苦瓜[19]、油菜[20]、桃金娘[21]和草莓[22]。所有这些物种的共同特征是每条染色体上只有一个着丝粒。换句话说,它们是单中心物种。此外,三个全中心物种的高质量基因组也已详细报道。三个罗蒙塔草属物种的所有全中心均匀分布在整个染色体上[34]。之后,豌豆染色体6的81.6 Mb着丝粒的新组装展示了另一种着丝粒组织类型,即元多中心着丝粒[35]。由于本研究组装的T2T参考基因组的可用性,在川桑中也鉴定出了真实的着丝粒m3cp重复和完整的着丝粒。
显著的是,川桑是一种具有多中心染色体的物种。据我们所知,之前没有报道过具有多中心染色体的物种。如上所述,单中心物种的基因组特征沿染色体长度显示出不均匀的分布模式。例如,基因通常集中在端粒区域附近,而重复序列通常聚集在着丝粒区域附近[11]。如报道的全中心染色体,三个罗蒙塔草属物种的所有全中心均匀分布在整个染色体上,主要基因组特征也沿整个染色体呈现均匀分布模式[34]。关于元多中心着丝粒,在染色体中发现了一个延长的主要收缩区,并在延长的主要收缩区中发现了多个独立的着丝粒区域[35]。相比之下,在川桑中,主要的基因组特征可以沿所有六条染色体分为几个区间。
考虑到不同类型着丝粒的分布模式,我们假设多中心染色体代表单中心和全中心染色体之间的一种中间状态。先前的研究表明,元多中心着丝粒染色体也代表单中心和全中心染色体之间的一种中间状态[35, 36]。与豌豆中的元多中心着丝粒染色体相比,其中一个主要收缩区有多个独立的着丝粒区域[35],我们还发现川桑中在一个着丝粒区域内有多个CENH3区域。然而,在桑树的六条染色体上可以在多个位置识别出着丝粒区域。
我们还关注多中心形成的机制。如报道的水稻,在删除了部分旧着丝粒后,一个新的着丝粒区域在染色体8的附近基因组区域中出现[37]。为水稻提出的机制可能解释了部分相邻CENH3区域的新形成。节段重复是元多中心着丝粒中CENH3区域传播的原因[35]。此外,赫利特隆元素介导了着丝粒重复的转座,加速了全中心物种中基于重复的着丝粒染色质的传播[34]。考虑到在染色体中存在多个长着丝粒区域,彼此距离较远,我们假设LTR类转座子在多中心形成中起重要作用,这些转座子可以介导大片段或染色体重排的易位。着丝粒区域内的完整Copia和Gypsy元素的插入时间明显晚于着丝粒区域外,这表明这些元素在着丝粒区域内确实比在其他区域更活跃(图S12,请参阅在线补充材料)。然而,要揭示多中心形成的精确机制,仍需进一步研究。
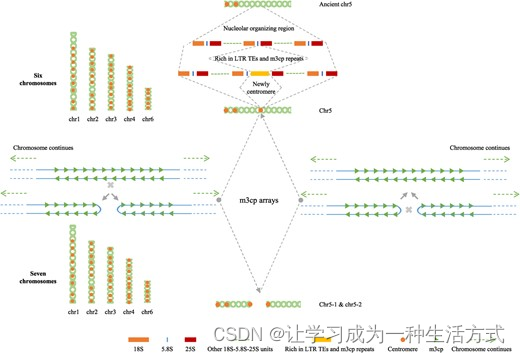
川桑染色体融合-裂变循环的拟议模型
一个需要解决的最有趣的问题是川桑中的染色体融合-裂变循环(图5)。在本研究中,染色体5表现出不同于川桑其他五条染色体的特定特征,接触图分析结果表明存在一个异常的染色质互作区域(chr5:28–32 Mb),指示了一个融合/裂变位点。值得强调的是,有两个区间显示出rRNA(18S、5.8S和25S)基因的聚集分布。通过细胞学实验发现了川桑中的染色体融合-裂变循环现象,FISH结果表明大致区域位于25S rRNA基因附近[5]。考虑到rRNA(18S、5.8S和25S)基因总是聚集在一个或多个染色体的核仁组织区(NORs)中[38],且在川桑的染色体5上仅标记了两个25S rRNA信号[5],我们提出这些rRNA(18S、5.8S和25S)基因都聚集在古老的染色体5上(图5)。如上所述,我们假设LTR逆转录转座子在多中心形成中起重要作用。由于在chr5的28.44-29.56 Mb区间内,LTR转座子的比例显著高于DNA转座子(P < 0.01),随着富含LTR转座子和m3cp重复的大片段插入古老染色体5的5.8S和25S rRNA基因之间,所有最初聚集在单一区域的rRNA基因拷贝被分成两个聚集区间。随后,由于该区域内高丰度的m3cp着丝粒重复,在m3cp阵列处形成了一个新的带有CENH3区域的着丝粒。在罗蒙塔草属物种中,着丝粒Tyba重复在染色体端对端融合中起重要作用[34],这一机制很好地解释了川桑中染色体数目从七条变为六条的变化。同时,由于丰富的m3cp着丝粒重复,其中单个m3cp重复包含超过95%的二聚对称序列,新形成的着丝粒也成为染色体裂变的热点。通过这种方式,确保了两个新形成的染色体各自拥有自己的着丝粒,从而维持了细胞周期的稳定。这里提出的模型不仅解释了新着丝粒的形成机制,还澄清了长期未解的桑树染色体融合-裂变循环的科学问题。
假设的的 Morus notabilis 染色体融合-裂变循环模型
材料与方法
植物材料和测序
在中国重庆北碚的桑树育种中心,桑树(Morus notabilis)在自然条件下栽培。采集新鲜的嫩叶组织并立即冷冻在液氮中,使用CTAB方法从新鲜的嫩叶中提取高质量的DNA,并使用NanoDrop One分光光度计(NanoDrop Technologies,Wilmington,DE,USA)进行评估。用Qubit 3.0荧光计(Life Technologies,Carlsbad,CA,USA)测量DNA数量。
对于SMRT测序,根据SMRTbell Express Template Prep Kit 2.0手册(Pacific Biosciences,CA,USA)制备标准HiFi文库。使用PacBio Sequel II系统(Pacific Biosciences,Menlo Park,CA,USA)测序川桑的基因组。对于ONT超长测序,使用SQK-ULK001试剂盒制备标准文库,并按照标准协议进行操作。使用PromethION测序仪(Oxford Nanopore Technologies,Oxford,UK)对纯化后的文库进行测序。对于HiC测序,使用Illumina HiSeq X Ten平台在150-bp PE模式下根据Illumina(Illumina,San Diego,CA,USA)的标准协议对基因组DNA进行深度测序。为了对川桑基因组进行适当的注释,从六种不同的组织(叶、根、芽、树皮、果实、花)中提取总RNA,使用RNAprep Pure Plant Kit(QIAGEN,北京,中国)。评估和测量后,根据Oxford Nanopore(SQK-PCS109)cDNA-PCR测序试剂盒的标准协议制备文库并使用PromethION测序仪进行测序。
基因组的de novo组装和质量评估
使用GenomeScope(v2.0)[39]对川桑的基因组大小进行k-mer频率分析估计,k-mer计数使用Jellyfish(v2.3.0)[40]。使用基于不同算法的多种策略进行基因组de novo组装,生成四组初级连锁体基因组。使用软件包CCS(v6.4.0, GitHub - PacificBiosciences/ccs: CCS: Generate Highly Accurate Single-Molecule Consensus Reads (HiFi Reads))按照默认参数过滤共识读数。对于HiFi测序,使用Hifiasm(v0.16.1)按照默认参数进行高质量HiFi读数组装以生成草稿基因组[25]。将从ONT超长测序获得的读数进行过滤,仅使用高质量读数进行进一步组装。使用NextDenovo(v2.5.0, GitHub - Nextomics/NextDenovo: Fast and accurate de novo assembler for long reads)进行组装,参数为“read_cutoff = 1k, blocksize = 1g, nextgraph_options = -a 1”。使用Necat(v0.0.1)[41]按照默认参数组装基因组。使用Flye(v2.9)进行组装,参数为“-i 3 -m 10000”。
初步组装后,使用Racon(v1.4.21)[42]和Pilon(v1.23)[43]结合ONT读数和Illumina PE读数进行三轮纠错和打磨,生成最终连锁体。使用fastp(v0.21.0)清洗所有Hi-C数据以生成高质量的清洁数据[44]。随后,使用HiCUP(v0.8.0, Babraham Bioinformatics - HiCUP Hi-C Analysis Pipeline)仅获得有效的Hi-C数据互作对,以进行染色体水平的组装。然后,使用ALLHiC(v0.9.8)[45]和Juicebox(v1.11.08)[46]对连锁体进行聚类、排序和定向。由Hifiasm生成的组装基因组被设为主干基因组,并使用ONT基因组填补主干基因组中的空隙。使用Winnowmap(v2.03)[47]将ONT中的端粒读数与基因组比对,并将获得的端粒序列(CCCTAAA/TTTAGGG)填补到缺少端粒的染色体中。随后,使用Winnowmap(v2.03)[47]再次将HiFi读数与基因组比对以进行纠错。最后,使用HiCExplorer(v3.6)[48]生成T2T基因组的基因组互作热图。
使用BUSCO(v5.2.2)结合胚植物_odb10数据库[49]评估川桑的T2T基因组的完整性。使用连锁体N50长度来揭示基因组的连续性,并评估基因组的准确性。使用BWA(v0.7.17)[50]将Illumina生成的所有读数映射到基因组。使用minimap2 [51]将PacBio HiFi和ONT读数映射到基因组,并计算LAI指数[52]以评估组装的基因组。
基因和重复序列的注释
使用两种策略识别重复序列:de novo和基于结构特征的策略。使用RepeatModeler(v2.0)[53]和EDTA(v2.1.0)[54]构建de novo重复库,使用RepeatMasker(v4.0.9)注释基因组中的散布重复。结合多种方法进行基因注释,包括ab initio、同源性基础和RNE-seq数据支持的方法。使用两个ab initio基因预测器,Augustus(v3.3.2)[55]和GlimmerHMM(v3.0.4)[56],并使用BUSCO(v5.2.2)[49]获取ab initio基因预测器的训练集。对于基于同源性的基因预测,我们选择了拟南芥[14]、川桑 [5, 7]、M. yunnanensis [7]、白桑[8]、印度桑[9]和大麻(Details - Cannabis_sativa_female - Ensembl Genomes 59),使用Exonerate(v2.2.0, GitHub - nathanweeks/exonerate: A fork of exonerate: a generic tool for sequence alignment)。
我们使用MAKER(v2.31.10)进行整个基因组注释流程,并使用NanoFilt(v2.8.0, GitHub - wdecoster/nanofilt: Filtering and trimming of long read sequencing data)、Pychopper(v2.7.2, GitHub - epi2me-labs/pychopper: cDNA read preprocessing)、Racon(v1.4.21)[42]和minimap2 [51]处理来自ONT平台的IsoSeq读数生成.bam文件。RNAseq读数通过fastp过滤,并使用hisat2(v2.1.0)[57]比对到我们组装的基因组生成其他.bam文件。随后,使用StringTie(v2.1.4)[58]和TransDecoder(v5.1.0, GitHub - TransDecoder/TransDecoder: TransDecoder source)预测基因模型。最后,使用MAKER(v2.31.10)[59]整合所有数据生成最终的基因模型集。
我们进一步按照以下方法预测基因功能:将所有编码基因搜索到其他数据库,包括KEGG [60]、InterProScan [61]、Swiss-Prot [62]、Pfam [63]和GO [64]数据库。使用tRNAscan-SE(v2.0.9)[65]识别tRNA基因,并使用RNAmmer(v1.2)[66]和INFERNAL(v1.1.4)[67]预测基因组中的rRNAs、snRNAs和miRNAs。
基因组和基因同源性
使用Mummer(v4.0.0beta2)[68]中实现的Nucmer功能在默认参数下进行基因组之间的比较。随后,使用参数‘-r -q’执行delta-filter命令以获得一对一的比对块区域。使用SyRI(v1.6)[69]识别这些基因组之间的同线性区域,并使用GenomeSyn(v1.2)[70]可视化基因组同线性。使用jcvi(v1.1.19)[71]中实现的MCscan pipeline的python版本在默认参数下进行和可视化基因同线性。
着丝粒和端粒序列的检测
使用seqkit(v2.3.1)[72]直接搜索端粒序列(5'-CCCTAAA/TTTAGGG-3')。通过Tandem Repeats Finder使用参数‘2 7 7 80 10 50 500’在基因组中识别串联重复。聚类这些重复序列后,我们计算了相应重复的丰度。连续且最丰富的重复单元被视为着丝粒串联重复。
染色质免疫共沉淀
ChIP的步骤如其他地方所述进行[74]。采集新鲜嫩叶并立即冷冻在液氮中。样品在4%甲醛中固定25分钟,超声处理染色质并进行质量控制。制备CENH3抗体(N端),对于ChIP,超声处理的染色质在4°C下与2 μg的CENH3抗体孵育过夜。随后,使用Dynabeads Protein G(Invitrogen,Carlsbad,CA,USA)捕获目标抗体,洗脱、解交联并沉淀结合的染色质。使用TruSeq ChIP文库制备试剂盒(Illumina,San Diego,CA,USA)制备ChIP-seq文库,并在Illumina NovaSeq6000上以150-bp PE模式测序。无抗体输入用作对照。
ChIP-seq分析和染色体上着丝粒的完整性
使用BLAST [75]将桑树中的m3cp卫星重复比对到每个参考染色体,参数为‘evalue = 1e-5, and –task blastn-short’。随后,使用BEDtools(v2.30.0)[76]合并所有相邻的m3cp单体,最大距离为50,000 bp,参数为‘-d 50 k’,以消除由于碎片化的m3cp阵列导致的间隙。然后,使用500 kb长的窗口再次扩展m3cp阵列的边界,直到没有找到m3cp重复。使用Cutadapt(v4.2)[77]修剪CENH3 ChIP-seq的原始测序读数,并使用BWA(v0.7.17)[50]将修剪后的读数比对到川桑基因组。过滤所有读数重复项以生成最终的BAM文件。使用deeptools(v3.5.1)中的bamCompare和bamCoverage工具将BAM文件转换为BIGWIG覆盖轨迹,参数为‘--binSize 50, --normalizeUsing RPKM’。使用MACS(v3.0.0b1)[79]通过比较ChIP和输入数据识别CENH3信号,参数为‘-broad -g -broad-cutoff 0.1’。随后,使用BEDtools(v2.30.0)[76]合并所有相邻的峰,最大距离为50,000 bp,参数为‘-d 50 k’,以消除由于碎片化的m3cp阵列或TEs插入导致的间隙。为过滤负面结果,仅保留超过五个峰的合并区域。此后,将边界扩展到两个侧翼方向的500 kb长的窗口,直到未找到ChIP-seq峰[11]。m3cp阵列和CENH3区域的边界用于定义着丝粒的边界。
甲基化分析
首先,使用Guppy(v6.0.1)生成纳米孔读数的基线调用,并按照推荐使用Tombo(v1.1.5, GitHub - nanoporetech/tombo: Tombo is a suite of tools primarily for the identification of modified nucleotides from raw nanopore sequencing data.)重新排列,创建索引并存储原始信号比对。随后,使用Tombo(v1.1.5, GitHub - nanoporetech/tombo: Tombo is a suite of tools primarily for the identification of modified nucleotides from raw nanopore sequencing data.)基于替代模型检测纳米孔读数中5mC(CG、CHG和CHH)的频率来分析胞嘧啶甲基化。通过甲基化读数数/无甲基化读数数计算胞嘧啶位置的甲基化频率。
荧光原位杂交
根据我们之前的报告[5]中描述的方法制备有丝分裂染色体。选择最佳的载玻片用于FISH。使用由擎科(北京,中国)合成的地高辛标记的m3cp寡核苷酸探针与川桑染色体杂交,并按照之前描述的方法进行FISH[5]。最终图像使用Adobe Photoshop CS6软件处理。

















 1695
1695

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









