










酶促4+2和2+2环加成反应:区域与立体选择性的理解与应用

01 有机合成
类似有机化学:狄尔斯–阿尔德反应
狄尔斯–阿尔德反应是[4+2]环加成反应中最具代表的,由共轭双烯与亲双烯体构建环己烯骨架的经典反应。反应有良好的立体、位置选择性。
该反应常作为合成环状化合物(特别是六元环)的首选战略,在诸多反应中它的实用性很显著。
1 反应机理
通常,带有给电子基的双烯体和带有吸电子基的亲双烯体进行反应。因为前沿轨道(双烯的HOMO和亲双烯体的LUMO)的能量差越小,能够使轨道相互作用而稳定,从而使反应更容易进行(通常电子要求型)。同理,亲双烯体带有给电子基,共轭双烯带吸电子基的反应也容易进行(反电子要求型)。

反应是按协同反应顺式加成来进行的,优先生成endo加成产物(endo规则)。通常电子要求型的情况下二次轨道相互作用大致能解释这一规则,但endo/exo选择性也受立体影响,根据不同底物,完全选择生成exo产物的例子也有报道。除此之外分子内的Diels-Alder反应由于环状结构固定,构型自由度较低,也不完全适用endo规则。

根据有机电子理论的推测,Diels-Alder反应的加成产物更容易使置换基处于邻位或对位(邻、对位规则)。具体可由前沿轨道理论来说明,即HOMO-LUMO的系数大的反应点易于相互重叠而加成。

环状过渡态的双烯是s-cis(cisoid)结构时可加成,s-trans(transoid)结构则不能发生Diels-Alder反应。如下反应中,Z-1,3-pentadiene很难转变为s-cis结构,反应性与E式相比明显低下。

02 化学合成
4+2和2+2环加成反应均是构筑环结构的重要有机化学反应,在复杂天然产物、手性药物的化学合成和生物合成方面有广泛的应用。发现、发展包括4+2和2+2在内的酶促环加成反应,是当前化学生物学研究的热点之一。近期,国际、国内研究团队相继报道了多个酶促4+2和2+2环加成反应,解析了环化酶的蛋白结构和催化机制,设计了新的环化酶,或通过定向进化实现了不同类型环加成反应的区域和立体选择性调控。相关研究为采用合成生物学的策略设计和优化新型环加成酶提供了理论基础和成功范例,有利于促进酶促反应在有机合成领域的应用。
环加成反应是将分子中两个或者更多的不饱和基团连接,同时形成两个新的碳-碳或碳-杂键,从而构筑环结构的有机化学反应[1]。环加成反应类型多样,包括4+2、2+2、6+4、1,3-偶极环加成等不同类型的反应。其中,4+2和2+2环加成反应受到广泛关注,在复杂天然产物和手性药物的合成中有广泛的应用。4+2环加成反应,根据催化机制可以分为协同和分步4+2环加成反应,其中协同的4+2环加成反应也称为Diels-Alder反应,通过具有共轭结构的双烯体与不饱和双键的亲双烯之间发生反应,产生六元环己烯或者碳-杂环己烯结构;而2+2环加成反应则是两个不饱和双键之间发生反应,产生四元环丁烷结构。4+2和2+2环加成反应被发现以来,经过近百年的不断发展,已经成为有机合成的有力工具,并且依旧是化学家关注的热点。随着蛋白质工程技术的发展,酶催化热潮的兴起,发现和发展条件温和、高效和高选择性的酶催化4+2和2+2环加成反应已然成为化学生物学家的重要研究方向。尽管许多天然产物的生物合成过程可能经历4+2或2+2环加成反应,但是证实的酶催化过程十分有限,而且都集中在4+2环加成反应[2-3],还没有酶促2+2环加成反应的报道,导致该类酶促反应的催化选择性机制不清楚,制约了人们对酶催化4+2和2+2环加成反应的理解及其应用场景的开发。
近期,国内外多个团队先后在不同天然产物的生物合成研究中发现了催化4+2和2+2环加成反应的新型酶,并解析了它们的催化和选择性机制,为调控和设计新型酶促4+2和2+2环加成反应以及拓展它们在合成化学中的应用提供了基础。本文将分别从酶催化4+2环加反应的研究进展和酶催化2+2环加反应的研究进展两个方面介绍上述工作,并阐述酶催化4+2和2+2环加成反应的选择性与调控机制。
1 酶催化4+2环加成反应的进展
从20世纪90年代开始,多种催化4+2环加成反应的人工酶被陆续报道,比如抗体酶、人工金属酶、核酸酶以及从头设计酶等,但是自然界是否存在催化协同4+2反应的Diels-Alder酶长久以来一直存在争议。尽管很早就报道Sol5、LovB和Macrophomate synthase等酶可能催化Diels-Alder反应,但是这些酶通常催化多步反应,难以进行机制研究,导致难以定论是否存在Diels-Alder酶。直到2011年,刘鸿文教授[4]在大环内酯天然产物Spinosyn A的生物合成中首次报道了能催化Diels-Alder反应的天然酶SpnF,尽管该反应能自发发生,但是SpnF的加入能大大提升反应的速率(约500倍),证实了Diels-Alder酶的存在;2015年,刘文团队在螺环乙酰乙酸内酰胺天然产物Pyrroindomycins(PYR)的生物合成中首次报道了两个完全酶依赖、单一构型的串联酶催化Diels-Alder反应,通过黄素依赖的环加成酶PyrE3和β-Barrel 桶状结构的环化酶PyrI4顺序地催化两个Diels-Alder反应,分别构筑萘环和[6,5]-螺环结构,形成PYR核心骨架[5],此后进一步通过结构生物学以及计算化学解析了环加成酶PyrE3和PyrI4的催化机制和选择性机制[6-9],彻底结束了关于自然界中是否存在Diels-Alder酶的争论,并开启了在天然产物生物合成途径中寻找4+2环加成酶的热潮。此后,国内外科学家陆续报道了多个不同类型的酶催化4+2环加成反应[10-16]。但是,大多数4+2环加成酶的催化机制和选择性机制仍不清楚,反应类型和酶的类型仍然有限。
近期,美国莱斯大学高雪教授、密歇根大学David H. Sherman教授和加利福尼亚大学洛杉矶分校K. N. Houk教授[17]合作,在含有特殊双环[2.2.2]二氮杂环辛烷结构的真菌天然产物21R-Citrinadin A(CTD-A)的生物合成研究中发现了一类以前被注释为NmrA转录因子的新型4+2环加成酶,它催化新颖的氧化还原过程介导的逆电子需求的氮杂4+2环加成反应(图1)。研究团队通过与CTD-A结构相似的天然产物生物合成途径中的4+2环加成酶为探针,找到了CTD-A的生物合成基因簇。但意外的是,探针基因对应的蛋白并不催化4+2环加成反应;相反,通过基因敲除确定了以前被注释为编码NmrA转录因子的ctdP基因与氮杂4+2环加成相关。作者通过化学合成获得了4+2环加成底物1,并利用体外生化测试确定CtdP是新型的4+2环加成酶,立体专一性地产生α-反式构型的氮杂4+2环加成产物2。值得注意的是,CtdP催化的4+2环加成过程是辅因子烟酰胺腺嘌呤二核苷酸磷酸(NADP+)依赖的,提示该催化途径涉及氧化还原过程。通过结构生物学、计算化学以及同位素标记试验,作者进一步揭示CtdP催化的4+2环加成反应涉及氧化、逆电子需求的氮杂4+2环加成以及还原三个步骤:首先,氧化过程产生4+2环加成反应必需的双烯体结构以及还原性辅因子;接着,催化核心的逆电子需求的氮杂4+2环加成反应,立体专一性地生成双环[2.2.2]二氮杂环辛烷核心骨架;最后,利用氧化步骤生成的还原性辅因子还原4+2产物中新生成的碳氮双键,完成双环骨架的构筑,并再生氧化型辅因子,完成一个催化循环。尽管上述过程涉及氧化还原和4+2环加成反应多个过程,但净结果是氧化还原电中性的,仅发生了氮杂4+2环加成反应,因此是一个新颖的氧化还原过程介导的氮杂4+2环加成反应。进一步,作者也对上述过程具体的催化和选择性机制展开了研究,通过分子动力学模拟(MD)和密度泛函理论(DFT)计算发现在环加成酶CtdP反应空腔中底物1的C16位与氧化型辅因子NADP+接近,通过NADP+拔取C16上的氢负离子形成还原性辅因子NADPH,同时生成含有亚胺双烯体结构的产物3,以发生逆电子需求的氮杂4+2环加成反应;在4+2环加成反应阶段,产生β-构型产物的过渡态能垒更低,可以自发发生,但酶能逆转其选择性,专一性地产生能量上不利的α-反式构型产物2,体现酶在立体控制中不可或缺的作用。
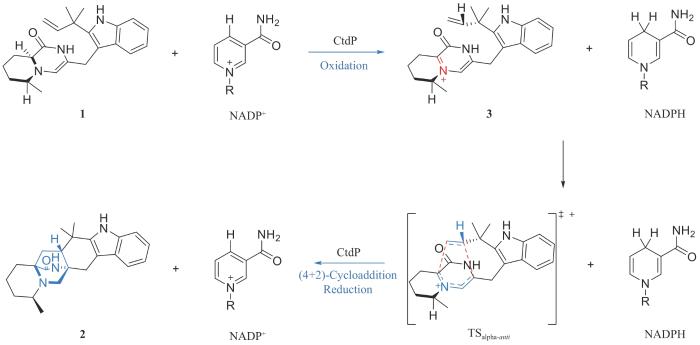
图1 CtdP催化的氧化还原过程介导的氮杂4+2环加成反应
不同于分子内4+2环加成酶的多样性,分子间的4+2环加成反应需要克服反应本身的能垒和分子靠近的反应熵,比分子内反应困难,发现的分子间4+2环加成酶也少很多。目前,仅有两个分子间Diels-Alder酶EupfF和MaDA被发现,还没有催化连续分子间Diels-Alder反应的报道。近期,中国医学科学院药物研究所胡友财研究员、中国科学院深圳先进院周佳海研究员和美国加利福尼亚大学洛杉矶分校K. N. Houk教授[18]合作,在真菌天然产物Eupenifeldin和Pycnidione的生物合成研究中首次报道了催化分子间串联Diels-Alder反应的酶EupfF和PycR1 (图2)。研究团队在2019年通过大肠杆菌异源表达了EupfF,发现它能催化单次分子间4+2环加成反应,生成三并环产物6,但是催化效率较低,始终无法合成结构更复杂的天然产物Eupenifeldin[19]。因此,研究团队从基因簇出发,利用真菌异源表达系统和底物饲喂实验,同时实现了Eupenifeldin及其结构类似物Pycnidione的组合生物合成,并推测EupfF及其同源蛋白PycR1实际上可以催化连续的分子间4+2环化反应,实现该类分子双吡喃环核心的构建。为了探究它们是否具有催化串联4+2环加成反应的能力,通过真核异源表达获得了EupfF和PycR1蛋白,分别将其与双烯体底物4与亲双烯体底物5反应,顺利生成了发生2次4+2环加成反应的天然产物Eupenifeldin和Pycnidione,由此确定了它们催化串联4+2环加成反应的新功能。值得注意的是,EupfF和PycR1发挥完整功能,依赖于对蛋白的糖基化后修饰。当功能蛋白没有发生糖基化后修饰时,仅发生一次分子间4+2环加成反应,生成三并环产物6和6′,并且活性较低;当发生糖基化后修饰以后,蛋白糖链可以促进蛋白质之间相互作用,形成二聚体,同时调节酶催化中心金属钙离子的络合,显著提升酶催化活性并赋予其更加完整的催化串联4+2环加成反应的能力。此后,研究团队进一步通过结构生物学和计算化学解析了两步4+2环加成反应的催化和立体选择性机制;并基于序列保守性分析和同源蛋白活性位点比对,通过定向进化实现了EupfF和PycR1第一步4+2环加成反应底物特异性的逆转以及串联4+2环加成反应步骤的控制。通过上述研究,增加了4+2环加成酶的类型和催化机制的多样性,为综合理解不同杂原子参与、不同电子需求的分子内或者分子间的4+2环加成反应的催化过程提供了基础。
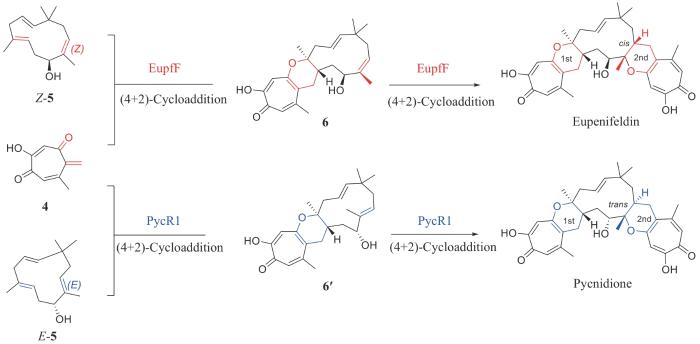
图2 EupfF和PycR1催化的分子间串联4+2环加成反应
2 酶催化2+2环加成反应的进展
2+2环加成反应能够高效构筑手性环丁烷,是合成科学重要的工具之一。尽管有很多催化2+2环加成反应的化学方法,但是酶催化的2+2环加成反应从未得到证实。近期,中国科学院上海有机化学研究所刘文团队、华中科技大学吴钰周教授团队和英国曼彻斯特大学Anthony P. Green团队分别从天然酶和人工设计酶两个方面证实了酶催化2+2环加成反应的可能性以及酶在2+2环加成反应选择性控制方面的优势。
利用协同2+2环加成反应必须在光激发条件下发生这一特性,吴钰周团队和Anthony P. Green团队[20-21]采用相似的理念设计了类似的催化2+2环加成反应的人工光酶(图3)。他们利用基因工程和蛋白质工程将合成化学发展的二苯甲酮类光敏剂通过基因密码子拓展技术定点插入到选定蛋白的手性空腔中,构建了含非天然催化活性中心的人工光酶。通过将三重态能量转移的光催化机制与蛋白质的精细超分子腔相结合,利用光敏单元控制反应,利用蛋白空腔控制手性和立体选择性,成功实现了人工光酶催化的2+2环加成反应。前者以吲哚衍生物为底物,以良好的底物普适性和手性选择性合成了环丁烷并吲哚啉类产物;后者以喹啉酮衍生物为底物,高效、高选择性获得了环丁烷并喹啉酮类产物。
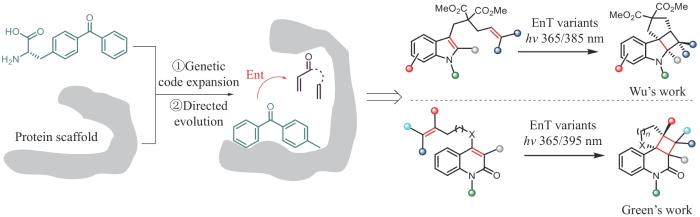
图3 人工光酶催化的2+2环加成反应
不同于利用光敏剂设计催化2+2环加成反应的人工酶,刘文团队与中国科学院上海有机化学研究所潘李锋研究员和美国加利福尼亚大学洛杉矶分校K. N. Houk教授[22]合作,从环加成酶的挖掘入手,在螺环乙酰乙酸内酯天然产物Pyrrolosporin A(PLO-A)的生物合成途径研究中发现了能催化2+2环加成反应的天然酶PloI4(图4)。研究团队从酶催化4+2环加成反应的立体选择性出发,对比研究具有endo-螺环结构的天然产物PLO-A和具有exo-螺环结构的天然产物Pyrroindomycins (PYR),以PYR生物合成途径中exo-4+2环加成酶PyrI4为探针,找到PLO-A生物合成途径中可能催化endo-4+2环加成反应的环加成酶PloI4。将PloI4与PYR生物合成途径中4+2环加成底物7进行体外活性测试,意外的是,不仅产生了预期的endo-4+2环加成产物8,还产生了exo-4+2环加成产物9以及exo-2+2环加成产物10。与人工光酶催化的2+2环加成反应不同,PloI4催化的2+2环加成反应不需要光参与,是完全非光激活的2+2环加成反应。进一步,研究团队利用结构生物学和计算化学解析了PloI4催化不同环加成反应,实现区域和立体选择性调控的分子机制,推导并验证了经由双自由基中间体分化产生热力学上有利的4+2和动力学上有利的2+2产物的反应过程。PloI4催化的2+2环加成反应与光酶催化的2+2环加成反应在机制上有本质的区别,前者是通过双自由基中间体分步方式实现,后者通过协同的周环过渡态实现。基于对机制的理解,研究团队通过定向进化,精准调控了该酶的化学和立体选择性,获得了分别催化exo-4+2、endo-4+2及exo-2+2单一环加成反应的突变蛋白,首次实现了将高阶的4+2环加成酶转变为低阶的2+2环加成酶。由此可见,通过对酶催化区域选择性的调控能实现不同类型环加成酶之间的转化,为通过酶与底物共进化的过程创造新的环加成酶提供了范例。
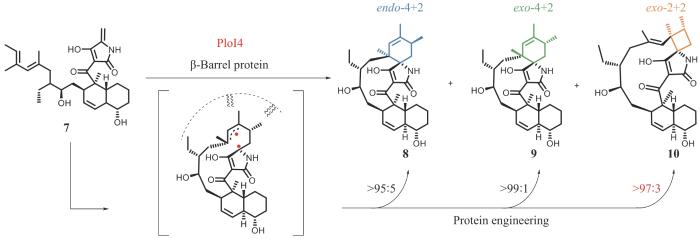
图4 PloI4催化的4+2和2+2环加成反应
作为合成环结构最有效的化学反应,4+2和2+2环加成反应具有广泛的应用空间,发现和发展催化4+2和2+2环加成反应的酶具有十分重要的意义。上述研究拓展了4+2和2+2环加成酶的多样性,加深了对酶催化4+2和2+2环加成反应的催化机制以及区域和立体选择机制的理解,为理性设计和调控酶催化环加成反应提供基础。尽管取得了突破性进展,酶催化4+2和2+2环加成反应领域仍然刚刚起步,发现的酶类型、底物类型仍然十分有限,制约了其应用。因此,仍需要继续发现和设计新型4+2和2+2环加成酶,拓展它们在合成化学中的应用。具体而言,可以利用生物信息学、化学生物、合成生物学等手段继续发掘天然的4+2和2+2环加成酶,特别是仍未有报道的天然途径中的2+2环加成酶基于机制解析,尤其是区域和立体选择性机制,设计和改造新型人工4+2和2+2环加成酶;开发具有底物谱宽泛4+2和2+2环加成酶催化剂,为绿色温和、高效、高选择性地合成环结构提供新的工具。
参考:
https://cn.chem-station.com/reactions/%e7%8e%af%e5%8c%96%e5%8f%8d%e5%ba%94/2014/02/post-515.html #有机化学
















 929
929

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









