






#################################################################
写在前面的话,需要会一些基本的linux指令
仅供参考https://www.quanxiaoha.com/linux-command/linux-shutdown.html
下面改一些文件需要会vim指令
仅供参考https://www.bilibili.com/video/BV1hy4y1z7tA/?spm_id_from=333.999.0.0&vd_source=c0e81733aa62a85725522f0de27627e4
#################################################################
一、加载intel编译器的环境
1、在bashrc里加入(一劳永逸)
if [ "$SSH_TTY" ]
then
source /opt/intel/oneapi/setvars.sh
fi2、命令行(每次打开新的ssh/终端都得输入一次)
source /opt/intel/oneapi/setvars.sh注:上述两种方式都可,正常显示
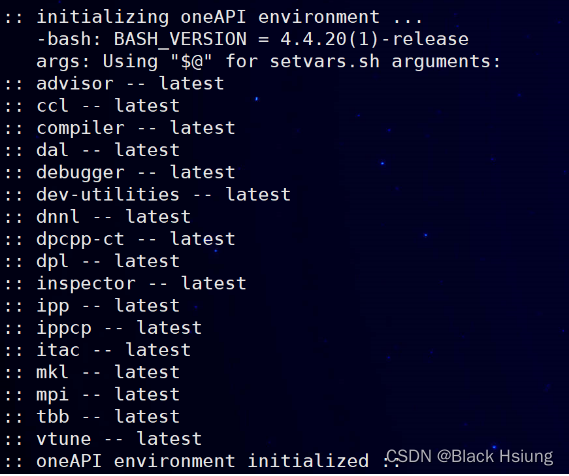
二、检查cuda-driver和cuda-toolkit环境
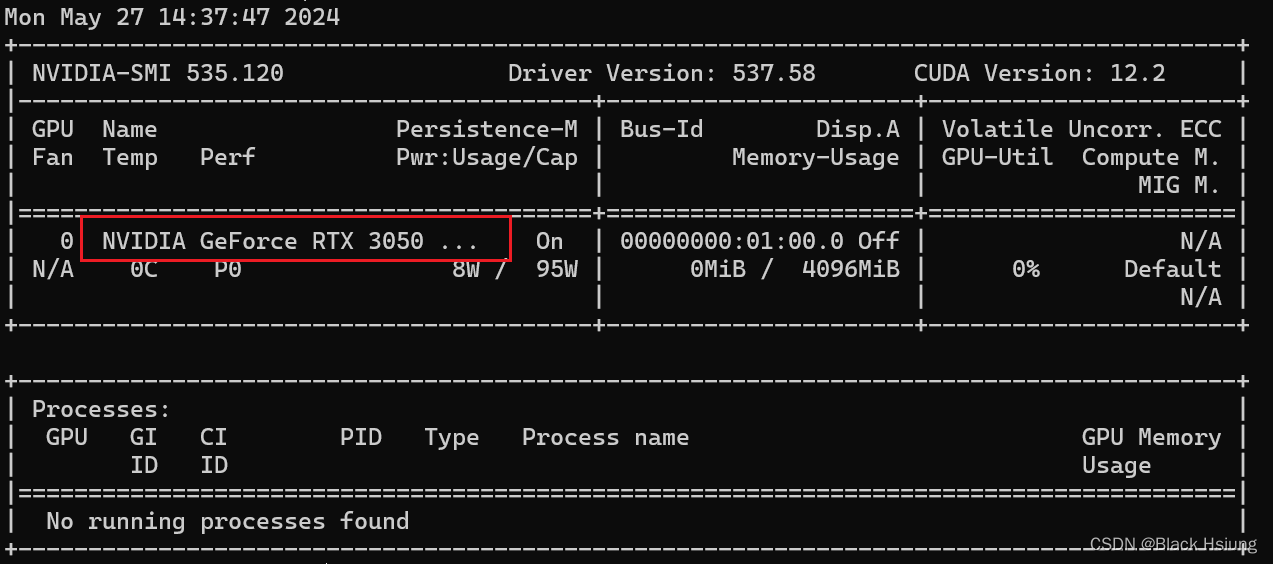
1、检查cuda-driver
nvidia-smi正常显示如下(此处需要看一下显卡型号,后面步骤需要用到)
非正常显示
bash: nvidia-smi: command not found...注:这是driver未安装或未正确安装,此时联系管理员(driver安装需root权限)
2、检查cuda-toolkit
bashrc加入或者命令行输入
export PATH=/usr/local/cuda/bin:$PATH
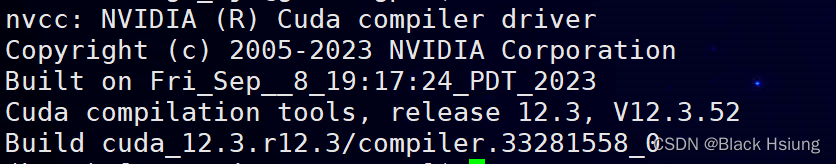
export LD_LIBRARY_PATH=/usr/local/cuda/lib64:$LD_LIBRARY_PATH检查路径是否正确,输入
nvcc -V正确显示
非正常显示
bash: nvcc: command not found...注:这是cuda-toolkit未安装或未正确安装,此时联系管理员(toolkit安装需root权限)
三、lammps-gpu安装
1、确保nvidia-smi和nvcc -V均正常
2、下载lammps
https://download.lammps.org/tars/index.html(建议年份比较新的版本)3、解压(这里以lammps-23Jun2022.tar.gz为例)
tar -zxvf lammps-23Jun2022.tar.gz4、编译lammps的gpu库
(1)cd lammps-23Jun2022/lib/gpu
(2)修改Makefile.linux主要两个地方
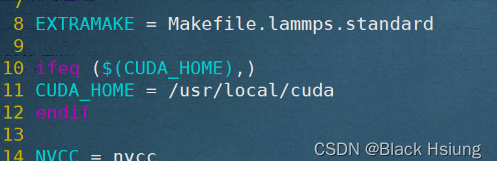
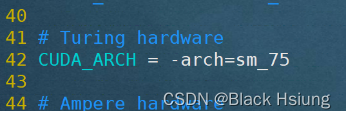
/usr/local/cuda替换成你的cuda-toolkit位置
arch=sm_75数字替换成你的显卡的算力值(75=7.5*10)
算力查询地址:https://developer.nvidia.com/cuda-gpus
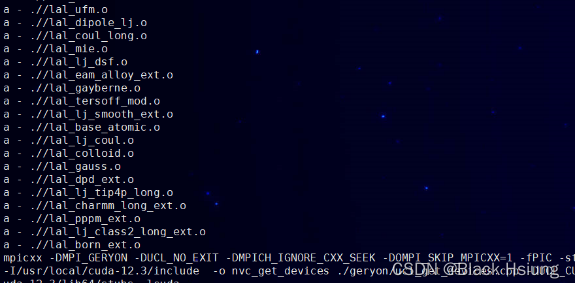
(3)make -f Makefile.linux(重新编译需要先make -f Makefile.linux clean)
编译成功显示
5、修改lammps的编译文件
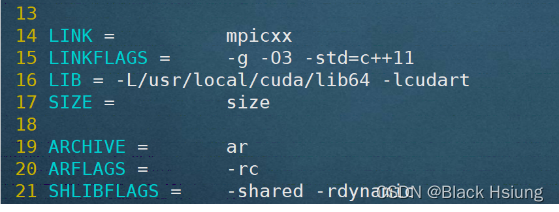
cd lammps-23Jun2022/src/MAKE
vim Makefile.mpi-L/usr/local/cuda/lib64替换成你自己的位置
6、编译安装lammps
cd lammps-23Jun2022/src安装必要的库,例如:make yes-gpu代表安装;make no-gpu代表卸载(库根据自己需要的任务自行安装)
注:可以使用make package-status检查自己安装的库(YES代表安装,NO代表未安装)
make mpi(重新编译需要make clean-all)
注:前一步中安装的库越多,make mpi时间越长,可以采用make mpi -j 4(4核同时编译,数字可改)7、ll lmp_mpi正常显示

四、测试
cd lammps-23Jun2022/examples/shear修改in.shear里的run的步数;随便改10000
### 纯cpu指令
mpirun -np 1 ../../src/lmp_mpi -in in.shear
### 加gpu指令
mpirun -np 1 ../../src/lmp_mpi -in in.shear -sf gpu -pk gpu 1对比运行的时间
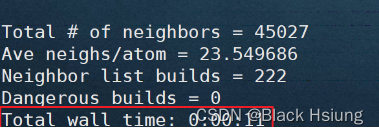
注:提交任务的指令有很多,建议到lammps官网(https://www.lammps.org/)或者手册查找(lammps-23Jun2022/doc/Manual.pdf)















 1785
1785

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









