







文献介绍

「文献题目」 Bridging the Gap between Connectome and Transcriptome
「研究团队」 Alex Fornito(蒙纳士大学)
「发表时间」 2018-11-16
「发表期刊」 Trends in Cognitive Sciences
「影响因子」 19.9
「DOI」 10.1016/j.tics.2018.10.005
摘要
最近构建的全脑基因表达图谱可以测量多个解剖位置数千个基因的转录活性,使得将基因表达的空间变化与连接组结构和功能的分布式特性联系起来成为可能。这些分析表明,基因表达的空间模式和神经元连接性密切相关,遵循广泛的空间梯度,跟踪微环路、区域间连接性和功能专业化的区域变化。叠加在这些梯度上的是基因表达和连接组拓扑之间更具体的关联,这些关联在不同物种和不同分辨率尺度上似乎是保守的。这些发现证明了全脑基因表达图谱在弥合健康和疾病中分子功能与大规模连接组组织之间的差距方面的实用性。
研究结果
1. 将分子功能与宏观大脑组织联系起来
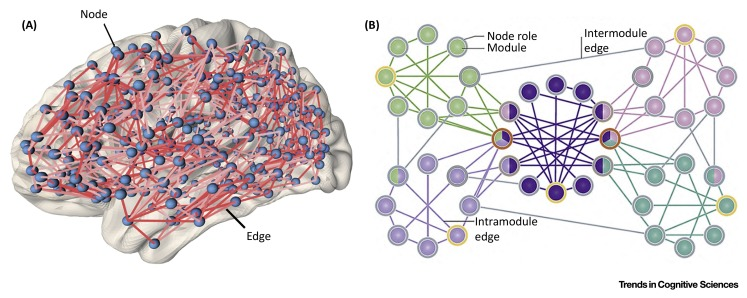
组成神经系统的全套元素和连接称为连接组。连接组可以表示为一个网络(network)或图(graph),其中每个神经元素是一个节点(node),元素对之间的每个连接是一条边(edge)(Box 1)。网络分析已被广泛用于表征从秀丽隐杆线虫到人类等物种中构建的连接组的特性,并在从单个神经元和突触到连接宏观大脑区域的白质束的范围内进行测量。这项工作表明,连接组具有复杂的「拓扑结构(complex topology)」。也就是说,它们的成对连接的特定模式「既不是完全规则的,也不是完全随机的」。并且许多拓扑特性在物种和尺度上是保守的(Box 1)。
Box 1 将大脑建模为复杂的网络
任何网络都可以表示为由边(edges)连接的节点(nodes)图。在连接组学中,节点可以代表神经元、神经元群体或宏观大脑区域,而边代表「结构连接」、「功能连接」或「有效连接」的某些指标(Figure IA)。网络中的完整连接可以用连接矩阵的形式概括,其形式相当于一个「图(graph)」。
通过将连接组提炼为其基本元素(节点和边),我们可以将大脑概念化为一类更广泛的复杂系统的一个例子。网络科学的数学基础以图论和统计物理学为基础,提供了丰富的测量方法来表征大脑网络拓扑和动力学的不同方面。
将网络科学应用于从不同物种和分辨率尺度获取的连接组数据,已经发现了拓扑特性的显着保守性。这些特性包括「经济布线(economical wiring)」,使得网络的总布线量相对于网络的拓扑复杂性而言接近最小;「分层模块化(hierarchical modularity)」和「小世界组织(small-world organisation)」;以及高度连接的「网络枢纽(network hubs)」的存在,这些网络枢纽也彼此紧密连接,形成一个「丰富俱乐部(rich club)」。枢纽之间的丰富俱乐部连接通常会延伸很长的解剖距离,连接不同的功能系统,并定位于调解大量网络流量,这表明这些昂贵的拓扑中心链接在整合大脑功能中发挥着核心作用。相比之下,模块化组织被认为支持专业化和功能分离。
Figure IB 显示出了具有丰富俱乐部(深紫色节点)和模块(由不同节点颜色表示;多色节点属于多个模块)的假设网络的拓扑投射。黄色节点边界标识省级枢纽(provincial hubs),它们在自己的模块内高度连接并支持功能专业化,橙色节点边界标识连接器枢纽(connector hubs),连接不同的模块。网络的该投射将具有相似连接分布的节点紧密地定位在一起(与 Figure IA 中的解剖投射相反)。
大脑网络本质上是定向的、加权的,并且具有异质节点(例如,大脑区域的微观结构特性不同,如细胞组成和细胞结构)和边(例如,连接可以是兴奋性的或抑制性的)。大脑网络也会随着时间而进化。迄今为止,大多数分析都依赖于最简单的网络表示:静态、无向的二进制图(Figure I)。改进的测量技术增强了我们解决大脑网络中的方向性、连接权重和异质性的能力,正在对连接组拓扑产生新的见解,并且对于提供与转录测定的发展相匹配的特异性水平非常重要。
连接组拓扑结构的这种显着保守归因于常见的选择压力,例如在促进复杂的自适应功能的同时最大限度地降低网络「布线成本(wiring costs)」的竞争驱动力。神经系统空间嵌入的物理和几何限制可以解释连接组拓扑的一些但不是全部方面,这表明遗传影响的额外作用。因此,人类双胞胎研究表明,大脑网络的拓扑特性,包括那些量化「网络经济(network economy)」特定方面的特性,是可遗传的。
我们如何识别导致连接组结构和功能变化的基因?在人类中,最常见的方法涉及测试给定大脑网络表型的个体间变异与 DNA 结构变异之间的关联,要么通过「候选变异(candidate variant)」研究,这需要有关基因的强有力的先验假设,要么通过「全基因组关联(genome-wide association)」,这需要大样本才能达到足够的统计功效。在小型模型物种中可以使用更广泛的方法,但这些方法通常很难扩展到高等哺乳动物。
在这里,作者考虑了一种将基因功能与整个大脑测量的神经表型联系起来的新方法的优点。这种方法利用全脑基因表达图谱,可量化许多不同解剖位置上数千个基因的转录活性(Figure 1)。这种基于图谱的方法的主要目标是了解基因表达的空间模式如何与大脑结构和/或功能的某些属性的空间变化相关。这一目标不同于行为遗传学和全基因组关联研究的传统重点,即了解基因如何影响个体的表型变异。全脑基因表达图谱的广泛解剖学和基因组覆盖使得全面绘制空间分布网络特性的分子相关性成为可能,从而有助于弥合大脑转录组和连接组之间的差距。
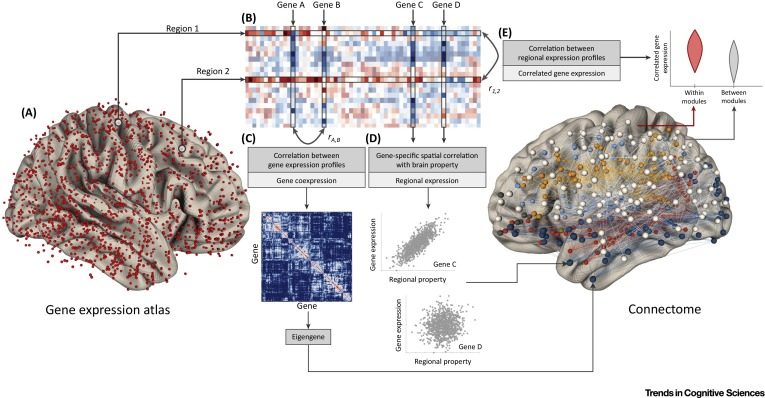
(A) 大脑表面显示取自六个供体大脑的不同皮质组织样本的位置,包括 Allen Human Brain Atlas(左)和使用扩散 MRI 数据构建的示例连接组(右),其中颜色表示不同拓扑模块的节点成员资格。
(B) 表达图谱中的每个样本都由跨基因表达值的向量来表征。这些区域特异性载体可以组合成区域×基因表达矩阵。
(C) 基因共表达是基因对的区域表达谱(即表达矩阵的列)之间的相关性。可以对每对基因进行估计以获得基因共表达矩阵。然后可以将该矩阵中的表达模式(例如特征基因)的汇总测量映射回大脑并与网络组织的区域属性相关。
(D) 区域表达模式的分析涉及分别提取每个基因的空间概况,并将其与某些大脑特性(例如体积、激活或节点度)的区域变化相关联。在这个假设的例子中,基因 C 显示出与大脑特性的空间相关性,而基因 D 则没有。
(E) 在相关基因表达 (CGE) 分析中,每个区域的基因表达向量(表达矩阵的行)彼此相关。然后,CGE 值可以与大脑结构或功能的成对测量相关联。在此假设示例中,将模块内连接的 CGE 值的分布与模块之间连接的 CGE 值进行比较。
作者首先概述与如何测量和分析基因表达相关的基本概念(Box 2)。然后,作者回顾了表明大脑转录结构由广泛的空间梯度主导的证据,这些梯度与细胞和环路结构的变化相关,然后再考虑叠加在这些梯度上的网络连接和拓扑的更具体的转录特征。总的来说,这项工作指出了基因表达的空间模式、神经元微环路、区域间连接以及大脑在健康和疾病中的功能特异性之间的密切联系(Box 2)。
Box 2 全脑基因表达图谱的临床应用
脑成像广泛用于绘制与特定临床疾病相关的神经结构和功能的变化,但导致这些宏观识别变化的分子机制通常尚不清楚。全脑基因表达图谱越来越多地被用来深入了解此类机制。
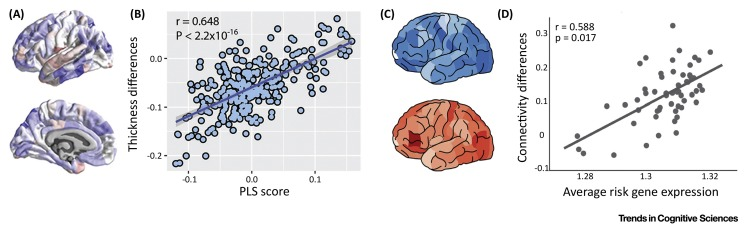
最通用的方法涉及测试某些成像表型中群体差异的空间拓扑与每个基因的空间表达模式之间的相关性。例如,「偏最小二乘(PLS)」分析已被用来识别遗传方差的组成部分以及与自闭症谱系障碍儿童的皮质厚度变化的拓扑相关的表达模式。Figure IA 显示了患者厚度变化的解剖学分布 (蓝色在对照中较厚;红色在自闭症中较厚) 并且 Figure IB 显示了这些变化与转录组方差的 PLS 分量之间的空间相关性,显示了 GO 类别突触传递的富集。类似的方法已被用来将基因表达与亨廷顿舞蹈病的结构连接性减少联系起来。在这两种疾病中,死后检查的患者大脑中发现许多与成像表型相关的基因表达发生了改变。这种对应关系表明,AHBA 转录模式与成像测量中病例对照差异之间的空间相关性可用于识别病理相关基因。
一种更有针对性的方法涉及使用表达图谱来阐明疾病风险基因的表型效应。在精神分裂症中,结构连接强度区域减少的空间模式与通过全基因组关联确定的疾病风险基因平均表达的空间模式相关。Figure IC 比较了 48 个精神分裂症风险基因平均表达的解剖学变化(上图;颜色越深表示表达量越高)与患者区域结构连接性的减少(下图;颜色越深表示患者减少程度越大)。两者之间的空间相关性如 Figure 1D 所示。在基因表达和皮质厚度变化之间没有观察到可比较的空间相关性,这表明这些基因对疾病易感性的影响是通过区域间连接而不是区域形态测量来介导的。在帕金森病中,tau 基因 MAPT 的空间表达模式与患者连接性降低的拓扑相关,而 SNCA(编码 α-突触核蛋白)则不然,这表明该疾病的宏观网络变化可能主要由 tau 介导病理。类似的分析已被用来了解淀粉样蛋白和 tau 蛋白如何影响阿尔茨海默病中不同的宏观大脑变化。
在第三种方法中,测试表达模式与规范成像表型相关的基因与给定疾病的已知风险基因的重叠。例如,一项研究确定了与健康青春期皮质厚度和髓鞘形成的发育变化相关的遗传变异的一个组成部分。该成分上的基因在与突触传递、谷氨酸能和离子通道信号传导相关的 GO 类别中富集,并且在精神分裂症的风险基因中过多。因此,这些发现建立了精神分裂症风险的遗传机制与正常大脑成熟之间的联系。
2. 绘制大脑转录图谱
构成个体 DNA 的核苷酸序列的变异会影响基因表达和蛋白质丰度,从而影响细胞功能并最终导致整个群体的表型变异。从 DNA 合成蛋白质是一个复杂的多阶段过程。就当前目的而言,两个最相关的阶段是转录和翻译。转录是基因表达的第一阶段,当 RNA 聚合酶读取解开的 DNA 片段以创建称为转录本的 cRNA 链时开始,转录本在蛋白质编码基因的情况下是 mRNA。当 mRNA 离开细胞核并到达核糖体核心时,翻译开始,转录物在核糖体核心被解码,合成氨基酸链,最终折叠成蛋白质。
基因表达通常通过 mRNA 水平间接测量,mRNA 水平指示基因的转录活性。然而,最终决定蛋白质丰度的是细胞的翻译活性。转录和翻译活性可以解耦,基因转录和蛋白质丰度之间的关系并不总是简单的。测量基因转录的优点是使用高通量检测更容易量化。
现有的转录图谱在所使用的表达分析、组织特异性和解剖覆盖范围方面各不相同。两个解剖学上最全面、使用最广泛的图谱是 Allen Mouse Brain Atlas (AMBA) 和 Allen Human Brain Atlas (AHBA)。AMBA 包括对每个组织切片中超过 19,000 个基因的基因表达进行原位杂交测量,这些基因通过细胞分辨率在全脑体积中每 200 μm 采集一次,而 AHBA 则包括对 3702 个区域中的 20,000 多个基因进行定量的微阵列测量不同的解剖位置和六个不同的大脑。
包含 AMBA、AHBA 和其他图谱的衡量标准均经过严格的质量控制程序,但在分析之前通常需要对数据进行进一步处理。每个处理阶段的选择都会影响最终结果,应谨慎做出。然后,通常在以下几个水平上分析转录组测量:(i) 「区域基因表达」;(ii) 「神经元件之间的相关基因表达(CGE)」;或 (iii) 「基因共表达网络」(Figure 1)。区域表达分析旨在确定基因表达的区域变异与某些区域特性之间的关联(例如「节点度 node degree」)(Figure 1D)。CGE 分析检查区域之间的转录耦合如何与网络组织的成对属性相关,例如两个区域是否连接,或者它们共享的连接类型(Figure 1E)。基因共表达网络的分析考虑了成对基因区域表达谱的相关性(Figure 1C)。因此,CGE 侧重于跨基因的区域-区域耦合,而基因共表达分析则侧重于跨区域的基因-基因耦合(Figure 1C、E)。请注意,该术语的使用并不一致,一些研究将 CGE 称为共表达,或将 CGE 矩阵称为转录脑网络。我们在这里使用这个特定的术语是为了尽量减少混乱。
基于图谱分析的一个共同目标是根据每个基因或基因对与给定连接组属性的空间或拓扑相关性对每个基因或基因对进行评分。然后使用统计推断来识别分数高于某个显着性阈值的基因。由于许多基因在功能上相关并且具有耦合的表达模式,因此通常使用富集统计测试在基因的功能类别水平而不是单个基因本身进行推断(例如,评估功能类别中的平均基因得分是否高于偶然的预期)。基因到功能注释的流行方法包括 「gene ontology (GO)」 和 「Kyoto Encyclopedia of Genes and Genomes (KEGG)」 。富集分析可以初步了解给定连接组特性的转录相关性,同时减轻分析数千个基因带来的多重比较负担。然而,GO 类别通常包含许多基因,因此需要进行后续调查来隔离单个基因的影响。
目前还不可能彻底检查基因表达的个体间差异,因为解剖学上最全面的图谱 AHBA 仅包含来自六个供体大脑的数据。因此,来自这些个体的表达测量通常与在独立的人群样本中量化的连接组特性相关。这种方法假设转录活性的区域差异大于个体间差异。一些初步证据支持这一假设,但目前我们对不同人的空间表达模式如何变化的理解是有限的。
尽管理解个体间变异的能力有限,但基于图谱的分析揭示了基因表达的空间模式如何与不同神经表型的解剖变异相关的重要见解。接下来我们考虑基因表达的广泛空间梯度如何跟踪区域微环路、宏观连接性和功能特异性的变化。
3. 空间梯度主导大脑的转录和连接结构
在全脑表达图谱中观察到的最显着的模式之一是强烈的空间自相关性,使得「物理上接近的区域具有更相似的转录谱」(即,它们在基因之间具有相似的表达值谱,其中相似性通常通过 CGE 量化)。这种基因表达的距离依赖性已在人类皮质、小鼠大脑、秀丽隐杆线虫头部中得到报道。它与神经元连接的类似距离依赖性相似,因此附近的神经元具有更高的连接概率(Figure 2A-D)。

(A-D) 在人类皮层中,(A) 相关基因表达 (CGE) 和 (B) 使用扩散 MRI 发现一对区域之间的联系的概率随着其间隔距离的变化而下降。红线代表拟合的指数趋势。通过顺行束追踪测量,小鼠大脑的区域间 (C) CGE 和 (D) 连接概率表现出类似的距离依赖性。
(E, F) 小鼠大脑区域的 2D umap 投射,基于 (E) 基因转录模式(4211 个基因,通过了我们的质量控制标准),以及 (F) 轴突连接模式。E 组和 F 组中的大脑区域以相同的方式着色。在两种表示中,具有相似解剖学划分的区域聚集在一起,在表达数据中最为突出。
(G) Allen Human Brain Atlas 中表达测量的前两个主要组成部分隔离了人脑中广泛的解剖学分区。左侧显示右侧使用的配色方案的解剖位置。
CGE 的距离依赖性和连接概率导致大脑大解剖分区内部和之间的转录组和连接组差异,并且可能是这些分区的功能和解剖专门化的基础 (Figure 2E-G)。在人类中,大解剖分区(例如,皮质与小脑)之间的转录差异大于分区内的差异(例如,不同皮质区域之间)。解剖分区内更细粒度的子区域拥有独特的转录特征的程度转录签名是一个悬而未决的问题;虽然有一些证据表明离散转录边界区分了海马和纹状体的功能细分,但在初级感觉运动区域之外的新皮质中很难识别这种边界 。新皮质中一个更一致的发现是表达模式遵循广泛的空间梯度。
在成人大脑中观察到的一个显着的基因表达梯度遵循「头尾轴(rostrocaudal axis)」,并且与产前观察到的类似转录梯度一致。这种空间模式可能与细胞结构的区域变化有关,内在环路和外在连接。皮质区域沿头尾轴显示神经元密度和数量的显着变化,因此「后部区域的神经元密度和数量都较高。这种梯度被前部区域神经元大小、轴突体积和棘数量增加的趋势所抵消,当一个神经元向前部移动时,会导致更复杂的连接」——无论是在局部微环路还是区域间连接层面(Figure 3A、B)。对于大脑较大的物种,前部和后部区域的皮质微观结构差异更大;被认为会导致大脑尺寸的种间差异;并可以解释人类在额叶介导的抽象认知过程中的优势。这些发现与人类 MRI 数据的双胞胎模型一致,显示遗传对新皮质表面积扩展的影响具有很强的头尾梯度,并且有证据表明越来越抽象的认知得到更多前部区域的支持。这些细胞组成和连接性的头尾变化被认为是由神经发生中相应且高度保守的空间梯度引起的,这导致后部区域神经元的产生延长,并最终导致密度增加;相反,前部区域神经发生的早期终止被认为能够延长突触连接的形成和巩固(Figure 3A,B)。
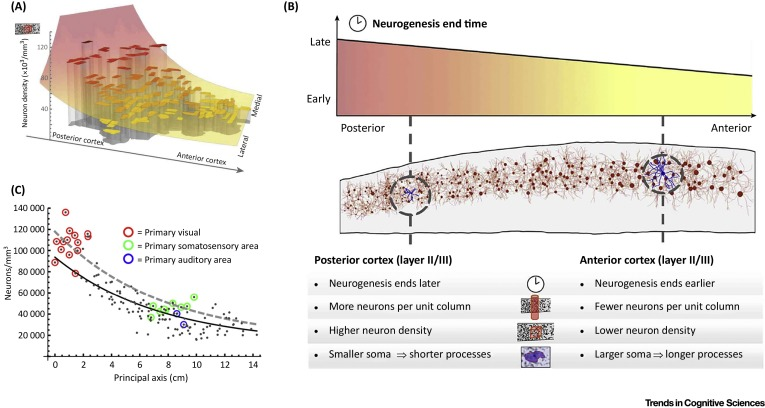
(A) 狒狒大脑中神经元密度的空间变化。条形代表取自不同组织样本的神经元密度测量值,位于沿头尾轴和中外侧轴。条形高度对应于每个样本的神经元密度。该表面代表定义神经元密度的全局趋势的数学函数,该神经元密度遵循从前外侧皮层延伸到后内侧皮层的轴。在猕猴和加拉戈猴中也观察到了类似的趋势。
(B) 哺乳动物大脑中沿头尾梯度变化的细胞和连接特性的总结。
(C) 狒狒皮层的区域神经元密度作为沿头尾轴位置的函数(较低的数字表示更多的尾部位置)。实线表示单因素数学模型的拟合,该模型仅表征沿该轴的变化。主要感觉区域(彩色圆圈)的密度始终高于模型预测。当使用考虑了头尾梯度以及初级和非初级皮层之间的区别(虚线)的双因素模型时,可以获得更好的拟合。
神经发生中的头尾变化的简单数学模型可以解释不同皮层区域和不同物种中神经元密度的皮层变化的许多方面,但包括初级和非初级皮层之间的额外区别的双因素模型更适合数据(Figure 3C)。主要区域和非主要区域之间的这种区别与众所周知的新皮质区域的处理层次结构一致,其范围从初级体感到高阶关联区域,并且被认为反映了刺激经历综合、多感觉处理程度的逐渐增加(Figure 4)。这种层次梯度在空间上与头尾轴相关(关联区域通常存在于额叶皮层),但其空间模式可能在关联皮层扩展的物种中有所不同,例如人类,其中层次梯度可能在定义细胞结构和连接性的区域差异。与这一观点一致,人类功能连接性的区域变异性的主要梯度密切跟踪皮质区域的功能层次结构(Figure 4D)。
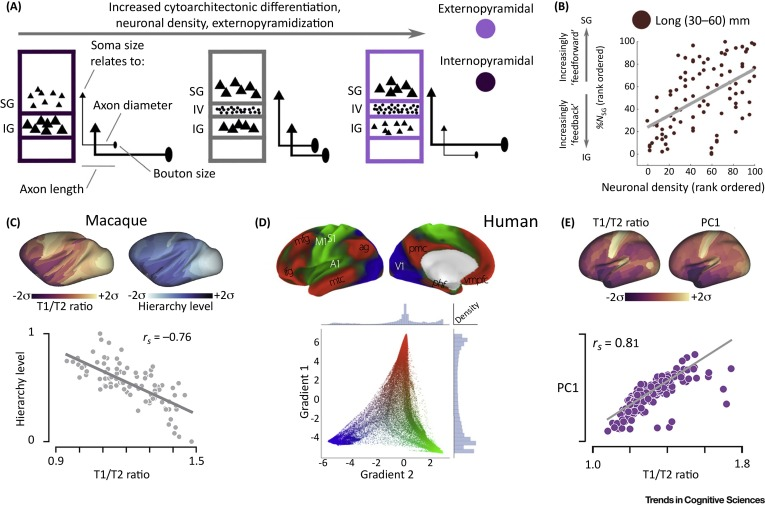
(A) 新皮质层次结构与细胞结构的众多变化相关。从关联到初级皮质的层次结构向下移动,与颗粒下神经元相比,颗粒上神经元有更大的细胞结构分化、更高的神经元密度和更大的胞体大小的趋势。胞体大小变化的这种趋势被称为外锥体化,伴随着平均轴突直径和突触布顿大小的相应变化。
(B) 从神经元密度较低的关联区域发出的长距离 (30–60 mm) 投射,按比例发送更多的反馈投射,通过逆行示踪剂 (%NSG) 标记的颗粒上层中的神经元百分比进行量化。
(C) T1-to-T2-weighted MRI 信号的比率跟踪髓磷脂含量的变化(左上),并与层次结构水平密切相关,使用从 %NSG 得出的测量值进行量化(右上、下)。
(D) 通过非线性降维确定的人类皮质功能连接的两个主要空间梯度。底部是一个散点图,位于由两个梯度定义的二维空间中的数千个皮质位点中的每一个。主梯度占大部分方差(梯度 1),将初级感觉运动区域(蓝色和绿色)与关联、边缘和旁边缘皮质(红色)分开。梯度 2 将视觉(蓝色)与躯体运动和听觉皮层(绿色)分开。顶部图像根据相同的梯度对解剖位置进行着色。
(E) 人类 T1/T2 MRI 比率的空间变化与大脑表达基因的第一主成分高度相关。顶部显示解剖对应关系,底部显示跨区域关联的散点图。
在猕猴中,皮层区域的层次排序是使用区域间投射的层状模式来定量定义的,观察到较低区域主要发送从上颗粒层到较高区域的第 4 层神经元的前馈投射,而较高阶区域主要发送源自原始的反馈连接位于颗粒下层中并且终止于较低区域中的第 4 层外部(Figure 4B)。因此,对小鼠、人类和非人灵长类动物的研究表明,跨皮质区域具有差异表达的基因往往表现出层特异性表达模式。基因表达的层状模式也与连接性密切相关,基因优先表达在第 3-5 层中,丰富了与轴突和神经元投射相关的 GO 类别;这一发现可能是由这些层中更高密度的远程锥体投射神经元驱动的。此外,区域专门化和区域间连接性的种间差异可能是由具有非保守层状表达谱的一小部分基因驱动的。与最后一点一致,与小鼠皮质相比,在人类第 2 层和第 3 层中表达丰富的 19 个基因(称为人类颗粒上富集或 HSE 基因)的 CGE 将感觉运动区域与边缘、旁边缘和联合皮质区分开来。与局部功能连接相比,具有更大远程连接的区域的这些基因的平均 CGE 更高,这是归因于关联皮层的整合功能的标志。因此,HSE 基因的空间模式表达被提议代表人脑中关联皮层相对扩展的转录标记。
基于层状连接性定义皮质层次结构的一个局限性是它需要侵入性测量。最近的一项研究发现,猕猴皮质的 T1-to-T2-weighted MRI 的对比度对皮质内髓磷脂含量敏感,与层状投射剖面推断的结构层次密切相关(Figure 4C),表明该比率提供了一种非侵入性方法来推断皮质区域的层次顺序。在人类中,作者报告了 T1/T2 比率与 AHBA 中第一主成分大脑特异性基因表达之间存在很强的空间相关性(Figure 4E)。已知在颗粒上层和颗粒下层中表达的基因显示出沿层次结构表达增加的趋势,而在第 4 层中表达的基因显示出沿层次结构表达减少的趋势。这些发现与高阶区域的皮质和胞体分化程度较低的证据相一致,特别是在颗粒下层,这被认为有助于更有效的远程连接(Figure 4A)。不同抑制细胞类型的转录标记、NMDA 接收亚基和调节受体也存在强层次梯度。与人类皮质中的这些结果一致,小鼠皮质的 T1/T2 比率在空间上与中间神经元细胞密度、细胞结构、长程轴突连接和大脑特异性基因表达的区域变化相关。小鼠和人类之间的直系同源基因的转录图谱显示出与两个物种的 T1/T2 比率相似的空间相关性,支持层次表达梯度的强烈保守性。
总之,这些链接连接组和转录组的全脑分析表明,基因表达的头尾和层次空间梯度、功能专业化和区域间连接密切相关。特别是,显示分层模式的基因往往具有层特异性表达模式,调节轴突形成和突触功能,并与不同的细胞和受体亚型相关,表明它们定义了皮质微环路和宏环路专业化的转录特征。
4. 基因表达携带有关成对大脑连接的信息
除了主导大脑转录景观的广泛空间梯度之外,基因表达和大脑连接之间是否还存在更具体的关系?Sperry 在 70 多年前进行的开创性实验发现了长距离解剖距离上轴突布线的显着特异性,这使他提出轴突通过连接源神经元和目标神经元的分子线索的精确规范来找到它们的目标:即所谓的「化学亲和性假设(chemoaffinity hypothesis)」。这种观点表明,成对连接的神经元件显示出耦合基因表达的高度组织模式。随后的大量工作集中于识别引导轴突与适当伙伴建立突触连接的特定分子,识别分子的重要作用,例如 netrins、slits、ephrins 和 semaphorins。其中一些和其他引导分子确实沿着梯度表达,尽管理论考虑和实验结果表明这些梯度只是几种不同线索的一类,这些线索在轴突导航到目标时通过发育而整合。
通过统计控制或从转录数据中删除这些趋势,或者通过构建适当的统计推断的空间受限零模型,研究了存在于广泛空间趋势之外的基因表达和连接性之间的关系。使用这些方法,结果表明,与未连接的节点对相比,大多数基因组的平均 CGE 更高,这是在小鼠的介观大脑区域水平上定义的。线虫的细胞水平和人类皮质形态协方差网络的宏观水平。导致小鼠连接区域 CGE 增加的基因在与轴突发育和维护、突触形成、功能、可塑性和细胞代谢。与线虫连接神经元 CGE 升高最密切相关的基因包括调节间隙连接、细胞粘附、迁移和轴突引导的基因。
其他几项研究调查了基因表达模式是否可以预测神经元件对之间连接的存在。在线虫中,经过训练的分类模型能够预测突触的存在使用 <300 个基因测量的表达模式,AUC 为 0.84,以及单个神经元的传入和传出连接伙伴,AUC 为 0.60。基因表达谱还可以预测中尺度小鼠连接组的区域间轴突连接模式,其准确性超过仅基于区域间距离的模型。对这些预测有很大贡献的基因在与突触和轴突形成相关的 GO 类别中富集。
总之,这些发现表明大脑转录组包含有关连接模式的信息,这些信息不能仅仅归因于广泛的空间趋势。这一结论与最近的一项研究一致,该研究报告称 CGE 结合空间距离和结构网络拓扑,最好地解释了小鼠大脑的成对功能连接,解释了 62% 的方差。主要注释为与钾和阳离子通道活性相关的 GO 类别的 568 个基因的子集对 CGE 的效果贡献最大。然而,区域间距离仍然是功能连接的最佳个体预测因子,能解释高达 55% 的方差。
在人类中,轴突连接性是通过扩散纤维束成像间接推断的,扩散纤维束成像已知影响长纤维束重建的空间偏差。因此,将纤维束描记测量与 AHBA 连接的结果不如秀丽隐杆线虫和小鼠的结果一致。一项研究发现 CGE 和成对结构连接性之间的关联性较弱,而后来的一项研究报告了种子区域结构连接性概况和特定特征基因之间的关联性较强。
功能连接性的研究不太容易受到扩散纤维束成像的偏差影响(但也有自己的偏差)。一项研究发现,具有高差异稳定性的基因的耦合表达与皮质区域更强的功能连接相关,并且具有高差异稳定性的基因在与轴突投射和引导相关的 GO 类别中得到富集。一项独立研究发现,使用 RNA 测序对 50 多个皮质样本进行量化的表达测量与自发低频功能 MRI (fMRI) 信号波动幅度的区域测量之间存在相关性,其中 26 个基因的子集解释了 10% 的信号幅度方差在默认模式网络的区域中。麻醉小鼠大脑的证据表明,自发的低频功能磁共振成像信号波动与某个区域的轴突输入总重量密切相关。因此,使用人类功能磁共振成像的这两项发现可能正在识别区域之间潜在结构连接的转录组相关性。
总之,虽然广泛的空间趋势可以解释结构连接性和相关转录活性的大部分差异,但连接性和基因表达共享特定信息,而这些信息不能仅通过其空间嵌入来解释。这些额外的信息可能由参与轴突引导和突触形成的一小部分基因携带,从而导致在基于图谱的成人大脑分析中观察到的富集特征。当考虑在发育早期阶段获得的表达数据时,一组扩展的基因是否与区域间连接相关仍然是一个悬而未决的问题。无论如何,连接特异性表达模式似乎补充了更广泛的空间趋势,例如在塑造连接组结构和功能方面定义沿头尾轴和层次梯度变化的趋势。
5.连接组拓扑的转录组特征
在跨物种和尺度观察到的拓扑特性的显着保守性(Box 1)表明其中一些特性,特别是那些不能归因于物理和几何限制的特性,受到遗传控制,因此应该具有独特的转录特征。这些特征在功能隔离(模块化架构)和集成(网络中心和丰富俱乐部连接)的拓扑基础方面得到了最广泛的研究(Box 1)。
第一项确定基因表达与网络组织之间联系的研究侧重于功能分离,证明与不同网络中的区域相比,人脑同一功能连接网络中的区域的 CGE 更高。驱动这种效应的基因在与离子通道功能相关的 GO 类别中富集,这些基因的结构变异导致了功能连接的个体差异。在人类形态测量协方差网络和线虫神经元连接组上定义的模块也发现同一网络内节点的 CGE 升高(Figure 5A、B)。虽然空间偏差可能有助于这些发现,但由于属于同一模块的节点往往在解剖学上共定位,它们可能无法完全解释这些影响。

(A) 人类区域×区域 MCN 和区域×区域 CGE 网络的模块具有相似的空间拓扑。不同的颜色表示模块从属关系。
(B) 秀丽隐杆线虫中所有神经元对的 CGE 值矩阵,使用均方列联系数进行量化。该矩阵已按神经元模块从属关系排序,模块边界由黑色方块描绘。对于同一拓扑模块内的连接神经元对(同一盒子中的神经元),平均 CGE 较高(深红色)。
(C) 示意图显示 hub 连接如何与小鼠大脑中的 CGE 相关。使用特定度阈值将节点分类为 hubs 或 nonhubs,从而允许在三种类型的连接之间进行拓扑区分:rich(两个 hubs 之间的链接)、feeder(hubs 和 nonhubs 之间的链接)、peripheral(两个 nonhubs 之间的链接)。然后可以从基因表达矩阵(右)中提取任何一对区域的相应表达向量来估计它们的 CGE。
(D) 顶部图显示小鼠区域间连接的度分布。该分布具有延长的尾部,与高度连接的 hubs 的存在一致。底部图显示 rich、feeder、peripheral 的 CGE,作为定义 hubs 的度阈值的函数。水平轴上的值越高表示 hubs 的定义越严格。灰色区域表示阈值制度,显示富裕俱乐部组织的拓扑证据(即中心之间的互连性比偶然预期的更高)。rich 链接的 CGE 急剧上升与这种拓扑富裕俱乐部政权的开始同时发生。水平虚线显示所有链路的平均 CGE。
(E) 与 D 组中所示的线虫连接组数据相同。与小鼠一样,CGE 在 rich 链接方面最高,并且在拓扑丰富俱乐部体系中急剧增加。
几项研究检查了拓扑整合的转录特征,主要关注高度连接的网络枢纽的作用。小鼠和大鼠大脑的早期工作确定了区域表达谱与节点度和「参与系数(participation coefficient)」之间的相关性,但没有报告表达数据中空间自相关校正的关联。在区域间转录耦合的水平上,一项研究拟合并消除了 CGE 值的距离依赖性(例如 Figure 2A),并发现这些距离校正的 CGE 值对于连接的网络中心对来说是最高的(称为 rich links),hubs 和 nonhubs 之间的链接处于中间(feeder links),连接的 nonhubs 最低(peripheral links)(Figure 5C,D)。平均而言,小鼠连接组的中心比其他区域对的间隔距离更长,因此它们升高的 CGE 与 CGE 随着区域间距离下降的总体趋势形成鲜明对比(Figure 2A)。驱动连接中枢 CGE 升高的基因富含与 ATP 氧化合成和代谢相关的 GO 类别,而与未连接区域相比,驱动连接区域 CGE 升高的基因,无论中枢状态如何,还富含与轴突和突触结构相关的类别和功能。代谢基因在这种假定的中枢连接转录特征中的强烈参与与证据相一致,即大脑网络中枢介导大脑中大部分信号流量,并且是连接组中代谢最活跃的元素之一。研究结果还表明,虽然调节轴突和突触结构和功能的基因表达可能区分连接区域和未连接区域,但成熟大脑中不同拓扑类别连接之间的主要转录区别可能与它们所连接区域的代谢需求有关。
最近,在线虫的微型连接组中也发现了连接的网络枢纽之间的 CGE 升高(Figure 5E)。这种效应不能归因于枢纽神经元之间在出生时间、谱系距离、模块从属关系、解剖位置、神经元亚型或分泌的神经递质方面的相似性。相反,这种效应归因于这样一个事实:线虫中的所有中枢神经元都对应于命令中间神经元的特定子类,这些子类在调节运动中发挥着关键作用,而运动是动物可实现的最复杂的行为之一。这些发现可能与哺乳动物的网络中枢主要位于关联皮层的证据相似,该皮层介导复杂的综合处理。
考虑到不同物种(无脊椎动物与哺乳动物)、空间尺度上的数量级差异(蠕虫中的单细胞与小鼠中尺度区域)以及表达数据的主要差异,蠕虫和小鼠之间连接的中心的 CGE 水平的一致性是值得注意的(从发表的文献中整理出的 948 个基因的二元表达数据,与小鼠中超过 17,000 个基因的定量原位杂交测量数据相比)。来自 fMRI 的证据表明,类似的特征也可能存在于人类中。具体来说,使用偏最小二乘法的分析确定了一个潜在成分,该成分描述了调节氧化代谢的基因中的转录变异,该变异与节点参与系数、模块间程度和远程连接相关。高水平是网络中心的特征。
总之,这些发现表明了在线虫、小鼠和人类中保守的模块化组织和中枢连接的转录特征。在哺乳动物中,驱动这些特征的基因似乎存在一些特异性,例如调节离子通道的基因可能会驱动功能连接网络内 CGE 升高,而调节氧化代谢的基因则主导中枢连接的转录特征。然而,目前尚不清楚大规模网络拓扑的这些明显特征是否真正不同。假定 hubs 是强互连的,并且可以被视为包括分布式模块本身,hubs 之间升高的 CGE 可以简单地代表 CGE 在功能相关和互连的神经系统内增加的趋势。这一结论与 HSE 基因与网络模块和 hubs 的转录特征有关的报道一致。考虑到皮层 hubs 主要存在于关联皮层中,CGE 和拓扑之间的关联也可能只是更一般原则的表现,例如分层组织。另一个相关和潜在相关的原则涉及具有相似层状结构和细胞结构的区域优先相互连接的明显趋势。由于与连接相关的基因通常表现出层特异性表达,我们可能期望 CGE 对于细胞结构相似的区域来说更高。为了测试这些可能性,需要对区域基因表达、细胞结构和连接性进行综合测量。
总结
这里回顾的证据表明,基因表达的解剖学变化与连接结构的变化密切相关。「这些变化主要由大规模空间梯度决定,其上叠加了更具体的网络连接和拓扑的转录特征」。将全脑表达测量与全连接组表型联系起来的第一波研究令人鼓舞,因为所检查的神经表型的转录相关性通常是合理的,并且暗示可能与神经元连接相关的基因。因此,他们为基于图谱的方法的实用性提供了初步支持,作为一种深入了解健康和疾病中空间变化的神经表型的转录相关性的方法(Box 2)。事实上,这些方法的优势在于它们可以轻松应用于人类成像数据并与在其他物种中构建的图谱相关。
基于图谱的分析的局限性之一是它们通常依赖于相关方法(Figure 1)。这种相关方法不能单独揭示给定神经表型的因果分子机制,但它可以优先考虑基因以供进一步研究。鉴于我们(目前)对基因如何影响大规模连接组特性的理解有限,优先顺序很重要。在模型物种中,可以使用更具侵入性的方法来询问优先级列表。在人类中,优先基因的等位基因变异可能与个体之间的表达数量性状位点和表型变异相关,从而识别影响基因转录和表型表达的特定 DNA 变异。通过基于图谱的方法对基因进行优先排序的优点是与单纯的全基因组搜索等位基因关联(约 20,000 个基因与约 100 万个变异)相比,多重比较负担更小。基于图谱的分析还可以阐明假定的疾病风险基因的表型效应(Box 2)。
目前可用的解剖学上最全面的转录组图谱测定了成人大脑中的表达,并且不描绘细胞特异性的表达模式。一些基因表达模式是新生的,但绝大多数(即>80%)在整个发育过程中表现出差异表达。发育解析转录组图谱的扩展及其与整个生命周期获取的成像数据的联系将揭示转录动力学如何与大脑网络成熟和衰老相关。单细胞转录组学的应用将增强此类工作,单细胞转录组学揭示了与细胞形态、生理学和连接性相关的基因组特征的巨大多样性。当大量组织样本中跨细胞类型的表达水平聚集时,这种多样性就会消失,就像在 AHBA 中一样。单细胞分析可扩展性的改进将产生具有前所未有的精度的新一代图谱。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布















 810
810











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









