







Alphafold2有docker和conda两种安装方式,官方提供的是docker版本的安装教程,conda版是民间修改版。
怀揣不信民间”歪门邪道“的想法,我原本更倾向于按照官方提供的流程进行部署,然而最后没有成功。。。很重要的一个原因是alphafold2本地部署的相关教程和讨论太少了,当然这也和它本身对硬件的要求有关系:一般的个人电脑完全带不起来。另外,一般人也就是用来预测一个结构,尽管使用colabfold的精度比alphafold2低,但是为了方便,大家应该都是能用colabfold就用colabfold了。
于是换赛道安装conda版,当安装成功后,conda版alphafold真香啊。。。
硬件:CPU:i9-13900k GPU:RTX 4090 内存:64G 硬盘:16T
系统&环境:Ubuntu 22.04.3 LTS,【gcc,g++,gfortran】10.5.0,CUDA 11.2,cmake 3.2.1,显卡驱动是安装好的-参考
软件:Anaconda3
一、Docker版安装(未成功)
首先按照官方提供的安装方式进行安装:
1、安装Docker(https://docs.docker.com/desktop/)如图1,并且可以以非sudo 权限(安装教程:https://docs.docker.com/engine/install/linux-postinstall/)运行
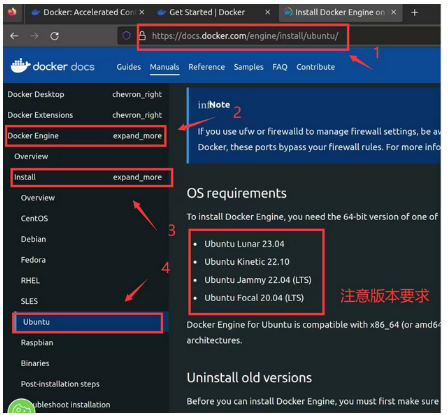
https://docs.docker.com/engine/install/ubuntu/
https://www.51cto.com/article/715086.html
2、Clone this repository and cd into it.
git clone https://github.com/deepmind/alphafold.git cd ./alphafold
3、下载序列文件
scripts/download_all_data.sh <DOWNLOAD_DIR> > download.log 2> download_all.log & 需要下载556G的文件数据,看scripts/download_all_data.sh 文件中的内容发现它是串行下载的((后面测试可以很容易改成并行下载)。在下载过程中很容易会报错误中断下载,但是好处是你重新下载的话,它会从中断的位置继续下载。 所以我是在下载中断的时候看下下载到哪一个文件了,然后将前面的注释掉,这样就能保证不会重复下载。
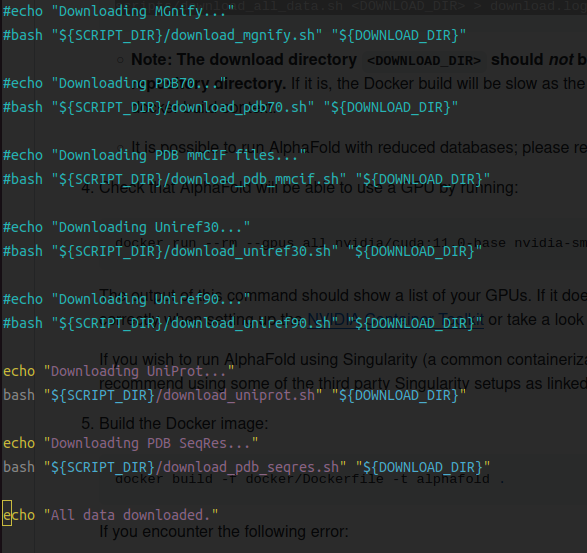
中间电脑显卡还出了问题送去检修了一段时间,断断续续的下载了20天左右(哈哈o(╥﹏╥)o)
下载结束后:
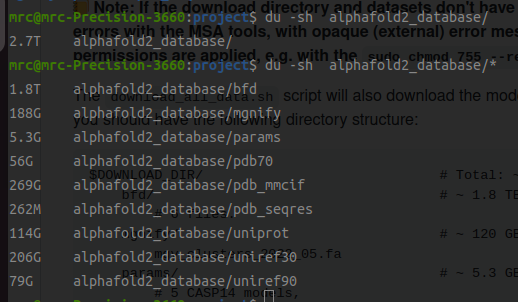
注意:默认下载的是BFD (1.8T) 而不是small BFD,如果磁盘空间不够,或者要求MSA速度快一些的时候(会牺牲部分精度),可以下载small BFD (解压后才17G)。
# small_bfd is a strict subset of BFD, generated by taking only the first non-consensus sequence from every cluster in BFD. 我这里选择将BFD和small_BFD都下载了,以针对不同的作业要求。(反正后者下载很快,也不占多少空间)
4、检查Alphafold可以使用GPU
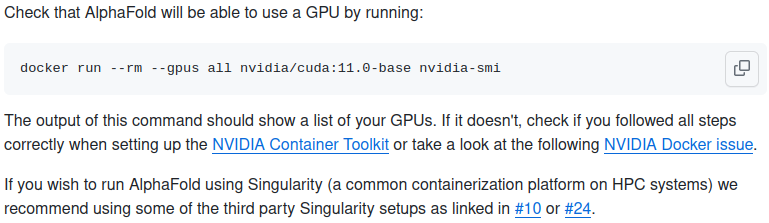
在我的配置下,使用的是:docker run --rm --gpus all nvidia/cuda:12.2.0-base-ubuntu22.04 nvidia-smi
结果报错:docker: Error response from daemon: could not select device driver "" with capabilities: [[gpu]].
这个报错表明Docker守护程序无法选则具有GPU能力的设备驱动程序,经检查发现是NVIDIA Container Toolkit未安装,于是点进去,安装:
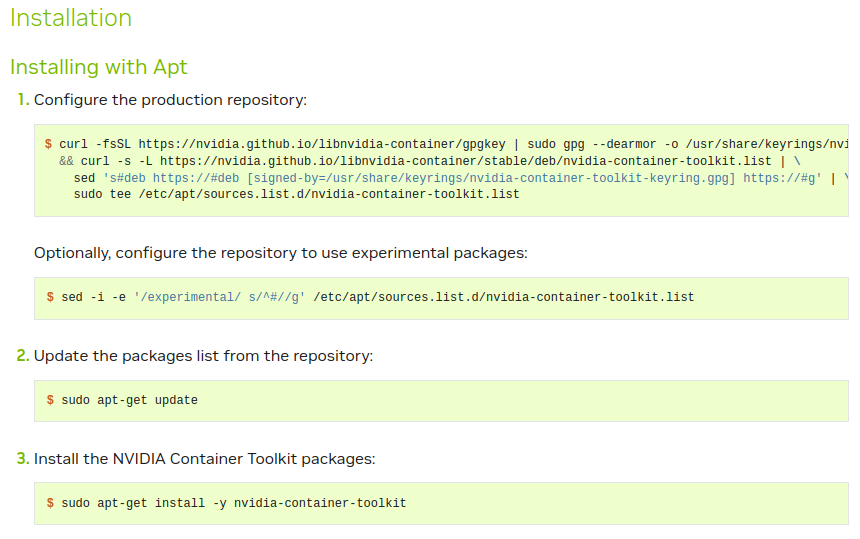
然后执行system restart docker,可以运行:
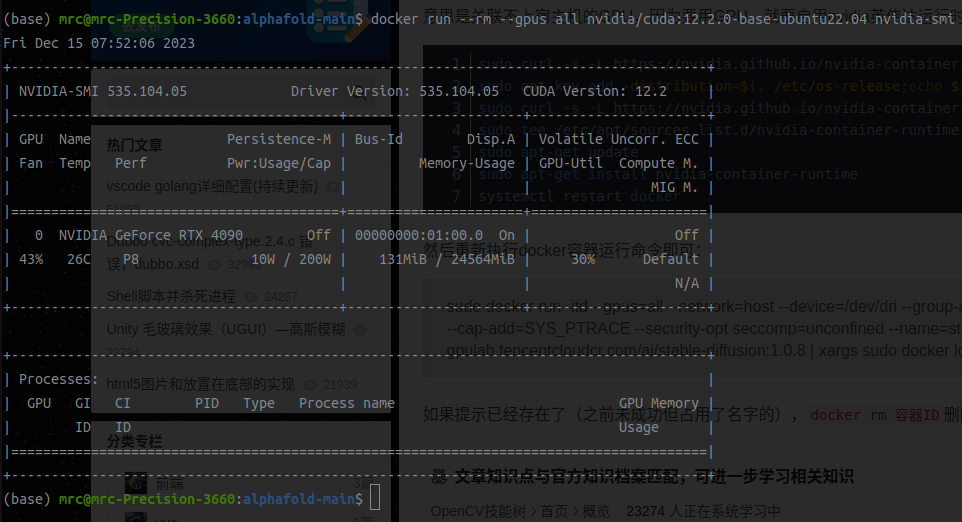
参考:https://blog.csdn.net/ys5773477/article/details/133642150
5、Build the Docker image:
在“/home/mrc/project/alphafold2/alphafold-main”下运行:
docker build -f docker/Dockerfile -t alphafold .
结果报错如下:


……试了很多种方法,这一步都未能完成
参考:
官方:https://github.com/google-deepmind/alphafold
https://www.bilibili.com/video/BV1Jm4y1V7JK/?vd_source=a9792134af400d0e2440e4ed09581d45
https://www.bilibili.com/read/cv26467969/
二、Conda版安装
使用docker安装在上述第5步一直卡着,于是开始尝试使用conda安装:
主要参考的安装流程:https://github.com/kalininalab/alphafold_non_docker 参考后安装失败的流程:https://github.com/kuixu/alphafold;https://github.com/google-deepmind/alphafold/issues/510
严格跟着上述教程走,即:
Create a new conda environment and update
conda create --name alphafold python==3.8 conda update -n base conda
Activate conda environment
conda activate alphafold
Install dependencies
- Change
cudatoolkit==11.2.2version if it is not supported in your system
conda install -y -c conda-forge openmm==7.5.1 cudatoolkit==11.2.2 pdbfixer conda install -y -c bioconda hmmer hhsuite==3.3.0 kalign2
- Change
jaxlib==0.3.25+cuda11.cudnn805version if this is not supported in your system
pip install absl-py==1.0.0 biopython==1.79 chex==0.0.7 dm-haiku==0.0.9 dm-tree==0.1.6 immutabledict==2.0.0 jax==0.3.25 ml-collections==0.1.0 numpy==1.21.6 pandas==1.3.4 protobuf==3.20.1 scipy==1.7.0 tensorflow-cpu==2.9.0 pip install --upgrade --no-cache-dir jax==0.3.25 jaxlib==0.3.25+cuda11.cudnn805 -f https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
Download alphafold release v2.3.1
wget https://github.com/deepmind/alphafold/archive/refs/tags/v2.3.1.tar.gz && tar -xzf v2.3.1.tar.gz && export alphafold_path="$(pwd)/alphafold-2.3.1"
Download chemical properties to the common folder
wget -q -P $alphafold_path/alphafold/common/ https://git.scicore.unibas.ch/schwede/openstructure/-/raw/7102c63615b64735c4941278d92b554ec94415f8/modules/mol/alg/src/stereo_chemical_props.txt
Apply OpenMM patch
# $alphafold_path variable is set to the alphafold git repo directory (absolute path) cd ~/anaconda3/envs/alphafold/lib/python3.8/site-packages/ && patch -p0 < $alphafold_path/docker/openmm.patch
Download all databases
……已经下载好
走完后直接运行:bash run_alphafold.sh -d ../../alphafold2_database/ -o ./dummy_test/ -f ../test.fasta -t 2020-05-14
报错:
(alphafold2) mrc@mrc-Precision-3660:alphafold-main$ bash run_alphafold.sh -d ../../alphafold2_database/ -o ./dummy_test/ -f ../test.fasta -t 2020-05-14
Traceback (most recent call last):
File "/home/mrc/project/alphafold2/alphafold-main/run_alphafold.py", line 41, in <module>
from alphafold.relax import relax
File "/home/mrc/project/alphafold2/alphafold-main/alphafold/relax/relax.py", line 18, in <module>
from alphafold.relax import amber_minimize
File "/home/mrc/project/alphafold2/alphafold-main/alphafold/relax/amber_minimize.py", line 25, in <module>
from alphafold.relax import cleanup
File "/home/mrc/project/alphafold2/alphafold-main/alphafold/relax/cleanup.py", line 23, in <module>
from openmm import app
ModuleNotFoundError: No module named 'openmm'
参考XXX,于是开始检查文件:(alphafold2) mrc@mrc-Precision-3660:alphafold-main$ vi /home/mrc/project/alphafold2/alphafold-main/alphafold/relax/cleanup.py
修改
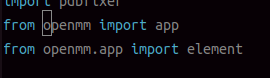
为
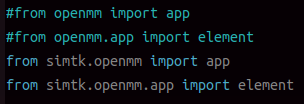
再次运行:
(alphafold2) mrc@mrc-Precision-3660:alphafold-main$ bash run_alphafold.sh -d ../../alphafold2_database/ -o ./dummy_test/ -f ../test.fasta -t 2020-05-14
Traceback (most recent call last):
File "/home/mrc/project/alphafold2/alphafold-main/run_alphafold.py", line 41, in <module>
from alphafold.relax import relax
File "/home/mrc/project/alphafold2/alphafold-main/alphafold/relax/relax.py", line 18, in <module>
from alphafold.relax import amber_minimize
File "/home/mrc/project/alphafold2/alphafold-main/alphafold/relax/amber_minimize.py", line 30, in <module>
import openmm
ModuleNotFoundError: No module named 'openmm'
继续修改:
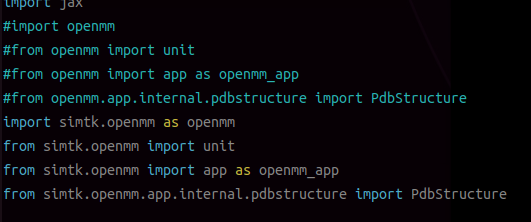
到这里,又有报错:
(alphafold2) mrc@mrc-Precision-3660:alphafold-main$ bash run_alphafold.sh -d ../../alphafold2_database/ -o ./dummy_test/ -f ../test.fasta -t 2020-05-14 -r none
FATAL Flags parsing error: Unknown command line flag 'run_relax'
Pass --helpshort or --helpfull to see help on flags.
google后发现有人和我有同样的问题:https://github.com/google-deepmind/alphafold/issues/811
原因是最新版alphafold2删除了run_relax,换成了- models_to_relax。这说明我用的是最新版。。。我明明从github作者处下载的是alphafold2.3.1,为什么现在用成了最新版呢?
……………………………………………………………………………………………………………………………………
这时候意识到,我的run_alphafold.sh文件放错地方了(放在了之前安装好的最新版alphafold里面),正确的应该放在从作者处https://github.com/kalininalab/alphafold_non_docker下载安装的
alphafold-2.3.1/路径下:

将run_alphafold.sh文件放到alphafold-2.3.1后,运行程序,成功执行!
然而运行过程中,在执行模型预测的时候出现报错,
I1218 22:38:50.427568 22891815339840 run_alphafold.py:191] Running model model_1_pred_0 on rcsb_pdb_1J01
I1218 22:38:51.968878 22891815339840 model.py:165] Running predict with shape(feat) = {'aatype': (4, 87), 'residue_index': (4, 87), 'seq_length': (4,), 'template_aatype': (4, 4, 87), 'template_all_atom_masks': (4, 4, 87, 37), 'template_all_atom_positions': (4, 4, 87, 37, 3), 'template_sum_probs': (4, 4, 1), 'is_distillation': (4,), 'seq_mask': (4, 87), 'msa_mask': (4, 508, 87), 'msa_row_mask': (4, 508), 'random_crop_to_size_seed': (4, 2), 'template_mask': (4, 4), 'template_pseudo_beta': (4, 4, 87, 3), 'template_pseudo_beta_mask': (4, 4, 87), 'atom14_atom_exists': (4, 87, 14), 'residx_atom14_to_atom37': (4, 87, 14), 'residx_atom37_to_atom14': (4, 87, 37), 'atom37_atom_exists': (4, 87, 37), 'extra_msa': (4, 5120, 87), 'extra_msa_mask': (4, 5120, 87), 'extra_msa_row_mask': (4, 5120), 'bert_mask': (4, 508, 87), 'true_msa': (4, 508, 87), 'extra_has_deletion': (4, 5120, 87), 'extra_deletion_value': (4, 5120, 87), 'msa_feat': (4, 508, 87, 49), 'target_feat': (4, 87, 22)}
2023-12-18 22:38:52.078354: W external/org_tensorflow/tensorflow/compiler/xla/stream_executor/gpu/asm_compiler.cc:231] Falling back to the CUDA driver for PTX compilation; ptxas does not support CC 8.9
2023-12-18 22:38:52.078381: W external/org_tensorflow/tensorflow/compiler/xla/stream_executor/gpu/asm_compiler.cc:234] Used ptxas at ptxas
2023-12-18 22:38:52.109243: E external/org_tensorflow/tensorflow/compiler/xla/stream_executor/cuda/cuda_driver.cc:628] failed to get PTX kernel "shift_right_logical" from module: CUDA_ERROR_NOT_FOUND: named symbol not found
2023-12-18 22:38:52.109323: E external/org_tensorflow/tensorflow/compiler/xla/pjrt/pjrt_stream_executor_client.cc:2153] Execution of replica 0 failed: INTERNAL: Could not find the corresponding function
Traceback (most recent call last):
File "/home/mrc/project/alphafold2/conda_version2/alphafold-2.3.1/run_alphafold.py", line 432, in <module>
app.run(main)
……
File "/home/mrc/software/Anaconda3/envs/Alphafold2/lib/python3.8/site-packages/jax/_src/dispatch.py", line 895, in _execute_compiled
out_flat = compiled.execute(in_flat)
jaxlib.xla_extension.XlaRuntimeError: INTERNAL: Could not find the corresponding function
在网上找到了这个链接 https://zhuanlan.zhihu.com/p/638287849,和当前这个报错一样,然而最后给出的解决方案却是Docker版的。。。
我尝试不使用GPU:bash run_alphafold.sh -d ../../alphafold2_database/ -o ./dummy_test/ -f ../test.fasta -t 2020-05-14 -g false
结果却可以正常运行,这说明是和GPU连接的问题。后续检查一下应该可以解决。
2023.12.19更新
google后发现前面的(报错:……jaxlib.xla_extension.XlaRuntimeError: INTERNAL: Could not find the corresponding function)问题应该和cuda版本有关(参考1,参考2 ),
于是,我将原来的cuda11.2直接换成12.2,GPU版可以正常使用了(只是使用GPU做relax会报错(可以试一下cuda11.3),然而这个无所谓,CPU优化也可以,并且更准确)。
运行代码参考以下的参数说明:
Running alphafold (v2.3.1):
Usage: run_alphafold.sh <OPTIONS>
Required Parameters:
-d <data_dir> Path to directory of supporting data
-o <output_dir> Path to a directory that will store the results.
-f <fasta_paths> Path to FASTA files containing sequences. If a FASTA file contains multiple sequences, then it will be folded as a multimer. To fold more sequences one after another, write the files separated by a comma
-t <max_template_date> Maximum template release date to consider (ISO-8601 format - i.e. YYYY-MM-DD). Important if folding historical test sets
Optional Parameters:
-g <use_gpu> Enable NVIDIA runtime to run with GPUs (default: true)
-r <run_relax> Whether to run the final relaxation step on the predicted models. Turning relax off might result in predictions with distracting stereochemical violations but might help in case you are having issues with the relaxation stage (default: true)
-e <enable_gpu_relax> Run relax on GPU if GPU is enabled (default: true)
-n <openmm_threads> OpenMM threads (default: all available cores)
-a <gpu_devices> Comma separated list of devices to pass to 'CUDA_VISIBLE_DEVICES' (default: 0)
-m <model_preset> Choose preset model configuration - the monomer model, the monomer model with extra ensembling, monomer model with pTM head, or multimer model (default: 'monomer')
-c <db_preset> Choose preset MSA database configuration - smaller genetic database config (reduced_dbs) or full genetic database config (full_dbs) (default: 'full_dbs')
-p <use_precomputed_msas> Whether to read MSAs that have been written to disk. WARNING: This will not check if the sequence, database or configuration have changed (default: 'false')
-l <num_multimer_predictions_per_model> How many predictions (each with a different random seed) will be generated per model. E.g. if this is 2 and there are 5 models then there will be 10 predictions per input. Note: this FLAG only applies if model_preset=multimer (default: 5)
-b <benchmark> Run multiple JAX model evaluations to obtain a timing that excludes the compilation time, which should be more indicative of the time required for inferencing many proteins (default: 'false')
使用GPU:
预测单体:bash ./run_alphafold.sh -d /home/mrc/project/alphafold2_database -f ../prediction_test/rcsb_pdb_1J01_monomer.fasta -o ../prediction_test/predict_results -t 2023-12-1 -e false
测试fasta:
>1J01_1|Chain A|test
ATTLKEAADGAGRDFGFALDPNRLSEAQYKAI
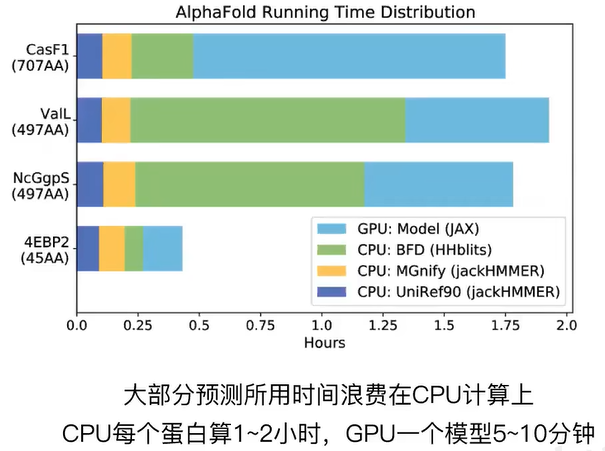
32分钟运行结束,主要时间花在CPU上。
##需要注意的是,run_alphafold.sh脚本必须在alphafold-2.3.1/文件夹下运行,因为它默认会调用该文件夹下的一些脚本和模块。
#如果对精度要求不高,可以使用-c reduced_dbs命令,调用small BFD数据库而不是BFD。 #测试了一个复合物序列,使用small BFD总时长可以缩短三分之一左右。
预测复合物:bash ./run_alphafold.sh -d /home/mrc/project/alphafold2_database -f ../prediction_test/rcsb_pdb_1J01_dimer.fasta -o ../prediction_test/predict_results/ -t 2023-12-1 -e false -m multimer -l 1
测试fasta:
>1J01_1|Chain A
ATTLKEAADGAGRDFGFALDPNRLSEAQYKAIADSEFNLVVAENAMKWDATEPSQNSFSFGAGDRVASYAADTGKELYGHTLVWHSQ
>1J01_1|Chain B
ATTLKEAADGAGRDFGFALDPNRLSEAQYKAIADSEFNLVVAENAMKWDATEPSQNSFSFGAGDRVASYAADTGKELYGHTLVWHSQ
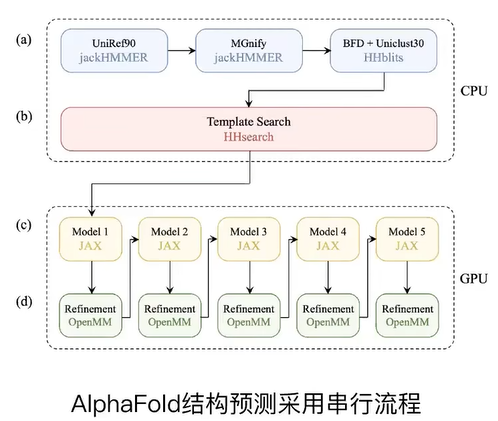
34分钟运行结束,主要时间花在CPU执行多序列比对(MSA)上(29分钟),GPU做结构预测的真正时间只有5分钟。如下图所示(From here1 and here2):

使用CPU:
在上述命令后加 -g false 即可,单独使用CPU,在模型预测部分的速度会慢很多。
结果解读:
1、输出的PDB中的B factor列中是每一个原子对应的pLDDT置信度预测结果,方便可视化
2、ranking_debug.json文件中提供了每个模型对应的ipTM (80%) + pTM (20%) 及其排名。其中,pTM强调蛋白复合体整体预测的准确性,ipTM强调蛋白质的交互界面预测的准确性
3、timings.json中是每一步运行的时间

2024.5.8更新
测试了allBFD,smallBFD以及colabfold在蛋白复合物(mCherry+intein)预测上的区别,结果为:三者基本一致,差别主要在于mCheery和ntein连接处的loop构型不一致,导致整体叠合效果一般。 但是如果只叠合mCheery部分,或者只叠合intein部分,基本能完全重合。
所以,如果是大批量预测,推荐使用smallBFD(速度快+可批量提交);如果是需要高精度,可用allBFD;如果是做测试或无硬件支持,直接用colabfold即可。
(版权所有,禁止一切转载!)















 5296
5296











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









