







 超级会员免费看
超级会员免费看

之前讲过临床模型预测的专栏,但那只是基础版本,下面我们以自噬相关基因为例子,模仿一篇五分文章,将图和代码复现出来,学会本专栏课程,可以具备发一篇五分左右文章的水平:
本专栏目录如下:
Figure 1:差异表达基因及预后基因筛选(图片仅供参考)
Figure 2. 生存分析,箱线图表达改变分析(图片仅供参考)
Figure 3. 基因富集分析(图片仅供参考)
Figure 4.构建临床预测模型(图片仅供参考)
Figure 5.训练集训练模型(图片仅供参考)
Figure 6.测试集测试模型(图片仅供参考)
Figure 7.外部数据集验证模型(图片仅供参考)
Figure 8.生存曲线鲁棒性分析(图片仅供参考)
FIgure 9.列线图构建,ROC分析,DCA分析(图片仅供参考)
Figure 10.机制及肿瘤免疫浸润(图片仅供参考)
########################### 下面是我们要做的图
Figure 1:差异表达基因及预后基因筛选(图片仅供参考)


### 我们准备的数据如下,上一节有讲到 ##

### 下面运行代码作图 ###
### 火山图 ###
此处我们的差异表达基因阈值设置为|LogFC|>0.5,adjusted.P.value < 0.05
setwd("C:\\Users\\ASUS\\Desktop\\自噬相关基因临床模型预测\\Figure 1")
dir()
data2 <- read.csv("TCGA_KIRC_diff_expression.csv",header = T,sep = ",")
data2$type <- ifelse((abs(data2$logFC)<0.5)|(data2$adj.P.Val>0.05),
"No alteration",ifelse(data2$logFC>=0.5,"Upregulated genes","Downregulated genes"))
head(data2)
levels(as.factor(data2$type))
data2
y = -log10(data2$adj.P.Val)
y
threshold<-as.factor(abs(data2$logFC)>=0.5 & data2$adj.P.Val<0.05)
library(ggplot2)
a <- ggplot(data2,aes(x=logFC,y=-log10(adj.P.Val),colour = type))+
geom_point(aes(x=logFC,y=-log10(adj.P.Val),colour = type),alpha=1, size= abs(data2$logFC))+
geom_vline(xintercept=c(-0.5,0.5),lty=4, col="grey",lwd=0.5)+
geom_hline(yintercept=-log10(0.05), lty=4, col="grey",lwd=0.5)+labs(x = "Log2(Fold Change)",y = "P Value (- log10)")
a
b <- a+scale_color_manual(values = c("dodgerblue2","grey40","coral2"))+
scale_fill_manual(values = c("dodgerblue2","grey40","coral2"))
b
c <- b+theme_bw()+labs(title = "TCGA-KIRC")
c
d <- c+theme(panel.grid.major = element_blank(),panel.grid.minor = element_blank())
e <- d+theme(plot.title = element_text(hjust = 0.5,size = 17,face = "bold"),axis.title.y.left = element_text(size = 17,face = "bold"),
axis.title.x.bottom = element_text(size = 17,face = "bold"))#### 设置纵坐标标题和主标题的大小,hjust设为0.5,表示将主标题居中。face = “bold”表示字体为粗体。
e
f <- e+theme(axis.text.x.bottom = element_text(size = 16,face = "bold",color = "black",vjust = 1,hjust = 1))
f
g <- f+theme(axis.text.y.left = element_text(size = 16,face = "bold",color = "black",vjust = 0.8,hjust = 0.6,angle = 90))
g
h <- g+theme(legend.text = element_text(size = 12,face = "bold"),legend.title = element_text(size = 14,face = "bold"))
h
### Venn图 ###
主要绘制差异表达基因和自噬相关基因的Venn图,我推荐以下网站进行绘制:
我们只要将差异表达基因和自噬相关基因的基因列表放入网站中:

我们先用代码将差异表达基因给导出来:
setwd("C:\\Users\\ASUS\\Desktop\\自噬相关基因临床模型预测\\Figure 1")
dir()
data2 <- read.csv("TCGA_KIRC_diff_expression.csv",header = T,sep = ",")
data2 <- data2[abs(data2$logFC)>0.5,]
data2 <- data2[data2$adj.P.Val<0.05,]
dim(data2)
write.csv(data2,"DEGs.csv")
然后将自噬和差异表达基因的基因列表上传:
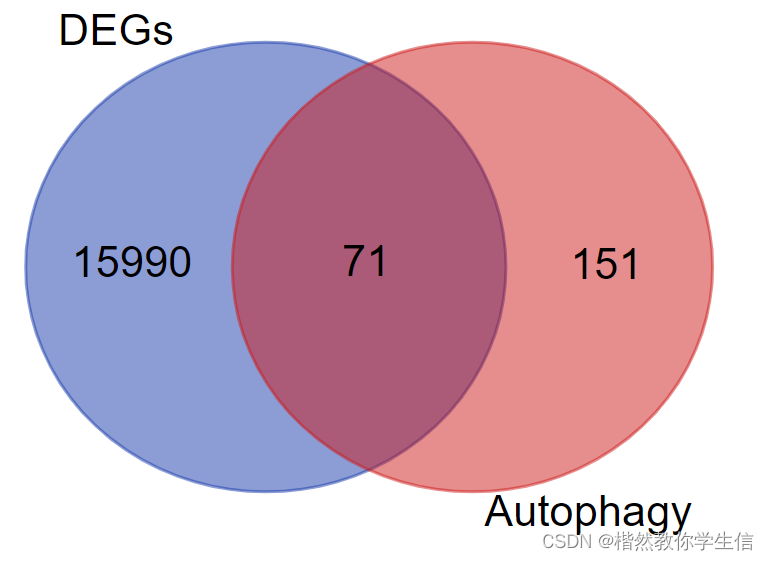
最终得到71个基因:

我们将用这71个基因绘制热图。
### 热图代码如下 ###
setwd("C:\\Users\\ASUS\\Desktop\\自噬相关基因临床模型预测\\Figure 1")
dir()
data2 <- read.csv("TCGA_KIRC_diff_expression.csv",header = T,sep = ",")
head(data2)
data2 <- data2[abs(data2$logFC)>0.5,]
data2 <- data2[data2$adj.P.Val<0.05,]
dim(data2)
write.csv(data2,"DEGs.csv")
head(data2)
auto <- read.csv("Autophagy.csv",header = T,sep = ",")
dim(auto)
auto[1:5,1:5]
library(limma)
rt=as.matrix(auto)
rownames(rt) <- rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data33=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
library(limma)
dim(data33)
data33[1:5,1:5]
data33=avereps(data33)
dim(data33)
data <- data33
data[1:5,1:5]
intersect(data2$X,auto$X)
data <- data[intersect(data2$X,auto$X),]
dim(data)
sample <- colnames(data)
sample <- as.data.frame(sample)
head(sample)
grep <- grep("^TCGA[.]([a-zA-Z0-9]{2})[.]([a-zA-Z0-9]{4})[.]([0][0-9][A-Z])",sample$sample)
length(grep)
grep
sample1 <- sample[grep,]
sample1 <- as.data.frame(sample1)
grep1 <- grep("^TCGA[.]([a-zA-Z0-9]{2})[.]([a-zA-Z0-9]{4})[.]([1][0-9][A-Z])",sample$sample)
length(grep1)
grep1
sample2 <- sample[grep1,]
sample2
sample2 <- as.data.frame(sample2)
head(sample1)
head(sample2)
names(sample2) <- "sample1"
sample <- rbind(sample1,sample2)
head(sample)
sample$cluster <- c(rep("Tumor",length(grep)),rep("Normal",length(grep1)))
head(sample)
dim(data)
data=data[,as.vector(sample$sample1)]
cluster=as.data.frame(sample[,2])
row.names(cluster)=sample[,1]
colnames(cluster)="Cluster"
rownames(sample) <- colnames(data)
library(pheatmap)
pheatmap(data, annotation=cluster,
color = colorRampPalette(c("dodgerblue3","dodgerblue2", "snow", "red","red2"))(50),
cluster_cols =F,
fontsize=8,
fontsize_row=8,
scale="row",
show_colnames=F,
fontsize_col=3)
所有的图片绘制完以后,均需要导出:

下一节开始匹配临床数据,绘制森林图。
如果看不懂代码的话,可以去学习之前的一个基础的专栏,或者最近出的R语言入门课程:
https://blog.csdn.net/weixin_46500027/category_11748288.html?spm=1001.2014.3001.5482![]() https://blog.csdn.net/weixin_46500027/category_11748288.html?spm=1001.2014.3001.5482https://download.csdn.net/course/detail/37644
https://blog.csdn.net/weixin_46500027/category_11748288.html?spm=1001.2014.3001.5482https://download.csdn.net/course/detail/37644![]() https://download.csdn.net/course/detail/37644
https://download.csdn.net/course/detail/37644
















 1820
1820











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











