






作者,Evil Genius
单细胞测序已成为在遗传学、转录组学和表观遗传学等不同水平上解开细胞群体异质性的有力技术,因此在基础研究和临床转化中具有深远的意义。细胞基因型主要通过单细胞DNA-seq (scDNA-seq)检测肿瘤中的体细胞突变,将细胞聚类成克隆并推断其进化动力学来研究。最近,越来越多的证据表明,在其他单细胞探针中,包括scATAC-seq和全长scRNA-seq(例如SMART-seq2),在核和线粒体基因组上也可以观察到体细胞突变的一个子集。另一方面,生殖系变异(又称单核苷酸多态性,SNPs)在单细胞测序数据中被更广泛地观察到,即使是在液滴为基础的平台上,如10XGenomics,这要归功于庞大的候选列表[在人群中约有700万个snp,频率为0.5 %]。种系snp不仅是perfect natural barcodes when multiplexing cells from multiple individuals,而且在通过细胞eQTL分析或等位基因特异性表达和拷贝数变化引起的等位基因失衡暗示功能调控方面也具有重要意义。
工具介绍
Cellsnp-lite是在C/ c++中实现的,并执行每个细胞基因分型,supporting both with (mode 1) and without (mode 2) given
SNPs。在后一种情况下,杂合snp将被自动检测。Cellsnp-lite适用于基于液滴的(例如10XGenomics数据)和well-based的平台(例如SMART-seq2数据)。
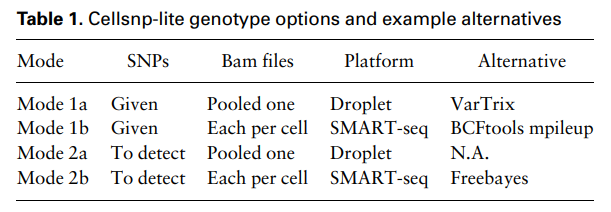
Cellsnp-lite需要以bam/sam/cram文件格式的对齐读取作为输入。细胞标签可以在多个bam文件(基于液滴的平台)中的细胞标签中进行编码,也可以由每个细胞bam文件(基于良好的平台)指定。这种灵活性还允许cellsnp-lite在bulk样品上无缝工作,例如bulk RNA-seq,只需将其视为基础良好的“细胞”。
pileup是在每个基因组位置进行的,对于给定的snp(模式1)或整个染色体(即模式2)。将获取覆盖查询位置的所有读取。默认情况下,丢弃那些低对齐质量的读取,包括MAPQ < 20,对齐长度< 30 nt和FLAG与UNMAP, SECONDARY, QCFAIL(和DUP,如果UMI不适用)。然后,对于基于液滴的样本(模式1a或2a),我们通过哈希图将所有这些读取分配到每个细胞中,或者对于well-based的细胞(模式1b或2b)直接分配。在每个cell中,计算所有A, C, G, T, N碱基的umi(如果存在)或读取。如果给定(即模式1),则从输入snp中取出REF和ALT等位基因,否则选择REF数量最高的碱基,ALT数量次之(模式2)。
当给定snp(模式1)时,cellsnp-lite将通过将输入snp按顺序等分入多个线程来执行并行计算。否则,在模式2中,cellsnp-lite将通过分裂列出的染色体并行计算,每条线程对应一条染色体。
在上述所有情况中,cellsnp-lite输出可选等位基因、深度(即REF和ALT等位基因)和其他等位基因的稀疏矩阵。如果添加参数' -genotype ', cellsnp-lite将使用表1所示的误差模型进行基因分型,并以VCF格式输出细胞作为样本。
安装
conda install cellsnp-lite
运行
Mode 1: pileup with given SNPs
Mode 1a: droplet-based single cells
Require: a single BAM/SAM/CRAM file, e.g., from CellRanger; a list of cell barcodes, e.g., barcodes.tsv file in the CellRanger directory, outs/filtered_gene_bc_matrices/; a VCF file for common SNPs. This mode is recommended comparing to mode 2, if a list of common SNP is known, e.g., human (see Candidate_SNPs)
cellsnp-lite -s $BAM -b $BARCODE -O $OUT_DIR -R $REGION_VCF -p 20 --minMAF 0.1 --minCOUNT 20 --gzip
As shown in the above command line, we recommend filtering SNPs with <20UMIs or <10% minor alleles for downstream donor deconvolution, by adding --minMAF 0.1 --minCOUNT 20.
Besides, special care needs to be taken when filtering PCR duplicates for scRNA-seq data by including DUP bit in exclFLAG, for the upstream pipeline may mark each extra read sharing the same CB/UMI pair as PCR duplicate, which will result in most variant data being lost. Due to the reason above, cellsnp-lite by default uses a non-DUP exclFLAG value to include PCR duplicates for scRNA-seq data when UMItag is turned on.
Mode 1b: well-based single cells or bulk
Require: one or multiple BAM/SAM/CRAM files (bulk or smart-seq), their according sample ids (optional), and a VCF file for a list of common SNPs. BAM/SAM/CRAM files can be input in comma separated way (-s) or in a list file (-S).
cellsnp-lite -s $BAM1,$BAM2 -I sample_id1,sample_id2 -O $OUT_DIR -R $REGION_VCF -p 20 --cellTAG None --UMItag None --gzip
cellsnp-lite -S $BAM_list_file -i sample_list_file -O $OUT_DIR -R $REGION_VCF -p 20 --cellTAG None --UMItag None --gzip
Mode 2: pileup whole chromosome(s) without given SNPs
For mode2, by default it runs on chr1 to 22 on human. For mouse, you need to specify it to 1,2,…,19 (replace the ellipsis).
This mode may output false positive SNPs, for example somatic variants or falses caused by RNA editing. These false SNPs are probably not consistent in all cells within one individual, hence confounding the demultiplexing. Nevertheless, for species, e.g., zebrafish, without a good list of common SNPs, this strategy is still worth a good try.
Mode 2a: droplet based single cells without given SNPs
# 10x sample with cell barcodes
cellsnp-lite -s $BAM -b $BARCODE -O $OUT_DIR -p 22 --minMAF 0.1 --minCOUNT 100 --gzip
Add --chrom if you only want to genotype specific chromosomes, e.g., 1,2, or chrMT.
Mode 2b: well-based single cells or bulk without SNPs
# a bulk sample without cell barcodes and UMI tag
cellsnp-lite -s $bulkBAM -I Sample0 -O $OUT_DIR -p 22 --minMAF 0.1 --minCOUNT 100 --cellTAG None --UMItag None --gzip
Output
cellsnp-lite outputs at least 5 files listed below (with --gzip):
-
cellSNP.base.vcf.gz: a VCF file listing genotyped SNPs and aggregated AD & DP infomation (without GT). -
cellSNP.samples.tsv: a TSV file listing cell barcodes or sample IDs. -
cellSNP.tag.AD.mtx: a file in “Matrix Market exchange formats”, containing the allele depths of the alternative (ALT) alleles. -
cellSNP.tag.DP.mtx: a file in “Matrix Market exchange formats”, containing the sum of allele depths of the reference and alternative alleles (REF + ALT). -
cellSNP.tag.OTH.mtx: a file in “Matrix Market exchange formats”, containing the sum of allele depths of all the alleles other than REF and ALT.
If --genotype option was specified, then cellsnp-lite would output the cellSNP.cells.vcf.gz file, a VCF file listing genotyped SNPs and AD & DP & genotype (GT) information for each cell or sample.
Full parameters
Usage: cellsnp-lite [options]
Options:
-s, --samFile STR Indexed sam/bam file(s), comma separated multiple samples.
Mode 1a & 2a: one sam/bam file with single cell.
Mode 1b & 2b: one or multiple bulk sam/bam files,
no barcodes needed, but sample ids and regionsVCF.
-S, --samFileList FILE A list file containing bam files, each per line, for Mode 1b & 2b.
-O, --outDir DIR Output directory for VCF and sparse matrices.
-R, --regionsVCF FILE A vcf file listing all candidate SNPs, for fetch each variants.
If None, pileup the genome. Needed for bulk samples.
-T, --targetsVCF FILE Similar as -R, but the next position is accessed by streaming rather
than indexing/jumping (like -T in samtools/bcftools mpileup).
-b, --barcodeFile FILE A plain file listing all effective cell barcode.
-i, --sampleList FILE A list file containing sample IDs, each per line.
-I, --sampleIDs STR Comma separated sample ids.
-V, --version Print software version and exit.
-h, --help Show this help message and exit.
Optional arguments:
--genotype If use, do genotyping in addition to counting.
--gzip If use, the output files will be zipped into BGZF format.
--printSkipSNPs If use, the SNPs skipped when loading VCF will be printed.
-p, --nproc INT Number of subprocesses [1]
-f, --refseq FILE Faidx indexed reference sequence file. If set, the real (genomic)
ref extracted from this file would be used for Mode 2 or for the
missing REFs in the input VCF for Mode 1.
--chrom STR The chromosomes to use, comma separated [1 to 22]
--cellTAG STR Tag for cell barcodes, turn off with None [CB]
--UMItag STR Tag for UMI: UB, Auto, None. For Auto mode, use UB if barcodes are inputted,
otherwise use None. None mode means no UMI but read counts [Auto]
--minCOUNT INT Minimum aggragated count [20]
--minMAF FLOAT Minimum minor allele frequency [0.00]
--doubletGL If use, keep doublet GT likelihood, i.e., GT=0.5 and GT=1.5.
Read filtering:
--inclFLAG STR|INT Required flags: skip reads with all mask bits unset []
--exclFLAG STR|INT Filter flags: skip reads with any mask bits set [UNMAP,SECONDARY,QCFAIL
(when use UMI) or UNMAP,SECONDARY,QCFAIL,DUP (otherwise)]
--minLEN INT Minimum mapped length for read filtering [30]
--minMAPQ INT Minimum MAPQ for read filtering [20]
--maxPILEUP INT Maximum pileup for one site of one file (including those filtered reads),
avoids excessive memory usage; 0 means highest possible value [0]
--maxDEPTH INT Maximum depth for one site of one file (excluding those filtered reads),
avoids excessive memory usage; 0 means highest possible value [0]
--countORPHAN If use, do not skip anomalous read pairs.
Note that the "--maxFLAG" option is now deprecated, please use "--inclFLAG" or "--exclFLAG"
instead. You can easily aggregate and convert the flag mask bits to an integer by refering to:
https://broadinstitute.github.io/picard/explain-flags.html
cellsnp-lite
生活很好,有你更好

















 681
681

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









