







–DOI: 10.1016/j.cell.2024.05.055
留意后续更新,请关注微信公众号:组学之心
研究团队和研究中心:

柏林医学系统生物学研究所–马克斯-德尔布吕克中心

:::
::::
Open-ST: High-resolution spatial transcriptomics in 3D
简介
- 空间转录组学(ST)方法与标准的单细胞方法区别主要在于,ST保留了捕获的转录组的2D空间坐标(可以直接观察细胞的排列及其在组织空间中的相互作用);避免了单细胞解离引入的偏差(单细胞解离会损耗某些细胞类型并激活应激途径)。
- 商业ST技术提供非靶向的转录组捕获,但成本较高且分辨率有限,包括Visium、CurioSeeker和Stereo-seq。探针法如CosMx空间分子成像仪、Molecular Cartography和Xenium原位成像仪,靶向预设计的基因组,不适合用于无偏发现或空间基因分型。
- 非商业ST技术则受限于低效捕获、低分辨率或实验设置繁琐复杂。
- 当前虽然存在细胞在3D中操作和通讯、构建功能组织和器官的情况,但还没有一个端到端的平台可以在3D中生成和计算分析ST。
- Open-ST旨在降低成本,结合高分辨率和高效的全转录组捕获,以及广泛的实验资源和开源软件,实现2D和3D数据处理和分析的无缝衔接(实现All in one)。
目前3D测序技术/3D基因组的发展正是热点,在这篇文章之前已有研究进行相关的探索,如’3D genomic mapping reveals multifocality of human pancreatic precancers’。抓紧时间补补知识,记录一下学内容。如有错误,请批评指正!
1.Open-ST的工作流程
文章的supplemental information有该测序步骤的介绍视频,感兴趣的小伙伴可以前去观看~
1.1 条形码配准和桥扩增
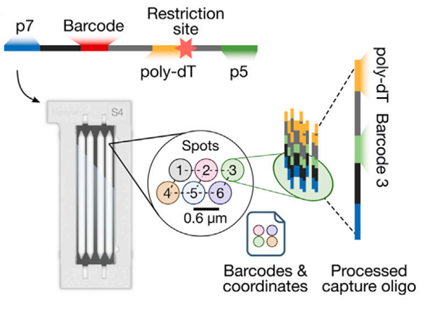
在Patterning Illumina Flow cell(此处为NovaSeq6000 S4)中对设计的寡核苷酸进行测序,可以在规则间隔的spot中配准条形码,对寡核苷酸进行处理以允许捕获多聚腺苷酸化RNA。
- ①条形码配准:通过测序寡核苷酸,在层流流动腔(Flow cell)上配准空间条形码序列及其相关的x和y坐标。寡核苷酸包括32-nt条形码、接头和poly(dT)区域。
- ②桥扩增:桥扩增技术用于生成密集堆积的spot,每个spot包含数千个具有独特条形码序列的克隆寡核苷酸。
- ③Flow cell使用:Open-ST采用NovaSeq6000 S4 Flow cell进行spot生成。纳米孔规则间隔,之间的中心到中心距离为0.6μm。
- ④与Non-patterned flow cells相比,Patterning flow cells可降低单个spot内混合条码信号的概率,并产生更高的spot密度。
1.2 精确切割RNA捕获区域

研究定制的3D打印设备可引导切割到所需尺寸的捕获区域。阴影网格表示用于Open-ST的Flow cell的成像区域,该Flow cell由四个7 mm宽的通道组成(成像6.3 mm)。
- ①条形码测序后处理:在条形码测序完成后,对寡核苷酸进行处理,使其能够捕获poly(A)转录本,并打开Flow cell。
- ②3D打印工具使用:我们定制的3D打印工具有助于将打开的Flow cell切割成小的捕获区域,同时防止表面划痕。
- ③捕获区域的尺寸选择:捕获区域的尺寸可以根据实验设计进行选择,最大尺寸为6.3 × 89 mm,受每个Flow cell通道的测序区域限制(图1C)。
- ④捕获区域的数量:每个3 × 4 mm单位可以形成360个捕获区域。
1.3 成像和测序
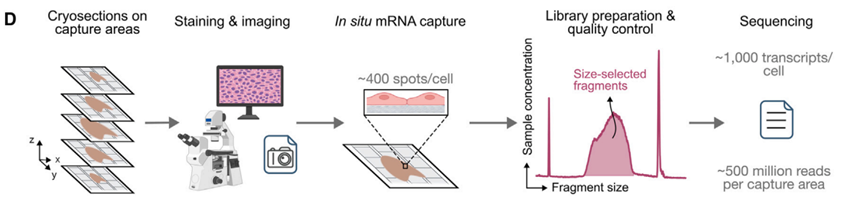
Open-ST允许从同一冷冻切片中同时分析组织形态(H&E)和空间转录组学(ST)。每个细胞覆盖大约400个测序纳米孔,检测出约1000个转录表达。
- ①RNA捕获:将胃蛋白酶和杂交缓冲液结合在一种溶液中,通过减少单链DNA和RNA分子的静电排斥来促进同时的组织通透和RNA捕获。
- ②透化条件优化:通过qPCR检测确定最佳透化条件,以实现最大程度的mRNA捕获。
- ③文库制备:在文库制备过程中,实施基于qPCR的质量控制,以确定最佳PCR循环数,避免文库扩增过多或不足。
1.4 组织形态学与ST测序数据的整合
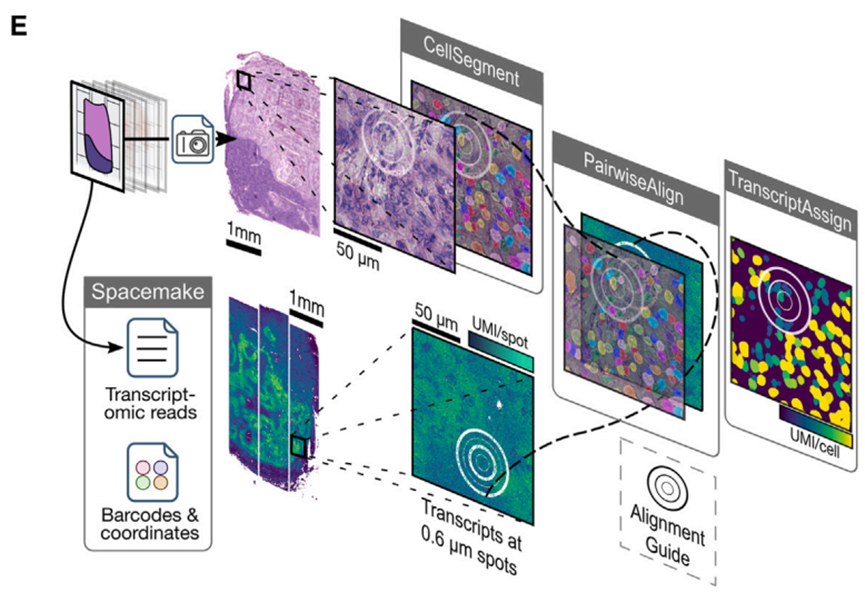
开源的openst软件包可以把组织形态学信息与对应的单个切片ST测序数据整合,包括自动细胞分割、模态成对比对和分割细胞中转录本的定量。
- ①图像预处理与细胞分割:原始H&E图像会自动进行预处理,并使用经过微调的Cellpose模型将其分割成单个细胞。细胞核分割后,细胞核边界的径向延伸增加了细胞质背景。
- ②转录组读数映射:使用第一次测序运行中的条形码坐标,使用Spacemake处理转录组读数并将其映射到组织空间中。
- ③模态成对比对:在成像和空间转录组模式中都可见的圆形标记会被自动检测出来,以进行无监督的成对比对,从而以1毫米的精度实现配准。
- ④分割和对比效果:分割和比对方法可以无偏地适应具有异质细胞大小和密度的组织,并自动从下游分析中排除没有细胞的区域。图像预处理和分割模型的微调提高了分割的精度,这通过与手动分割的基准测试得到证明。
- ⑤参数鲁棒性:该流程对于径向延伸距离、比对精度和其他参数的选择具有很强的鲁棒性;默认值在分割细胞数量、每个细胞的UMI和基因数量以及径向延伸时线粒体转录本的积累之间提供了平衡。
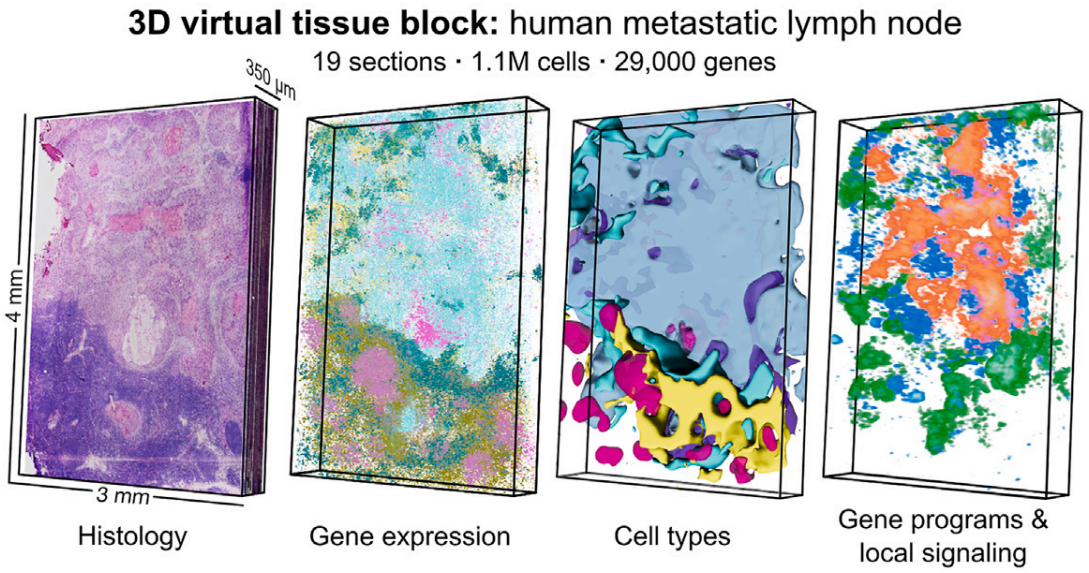
1.5 3D组织重建与虚拟组织块可视化
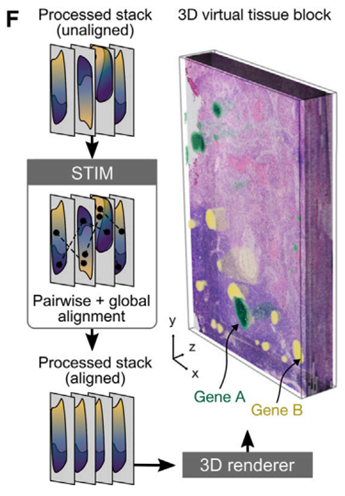
Open-ST 适用于任何组织的3D空间重建。研究重建了一个转移性淋巴结,整合了H&E染色和基因表达。使用空间转录组学成像框架(STIM)对齐连续切片,从而获得组织的3D表示。成像和转录组学模式都可以使用现有软件作为 3D 虚拟组织块同时进行查询,以实现科学可视化。
2.Open-ST平台捕获转录本的效果展示
研究将Open-ST应用于:胚胎小鼠头部、成年小鼠海马体、人类原发性肿瘤(HNSCC)以及与患者匹配的健康和转移性淋巴结。
所有样本中,大部分转录本(65%-78%)能够唯一地映射到基因组上,这意味着大多数读取的数据能够准确地定位到基因组的位置,而且核糖体RNA的占比小(2.5%-15.3%)。平均而言,55%映射到基因区域的读取被分配给来自第一次测序运行的空间条形码,说明超过一半的基因区域读取能与空间信息关联。
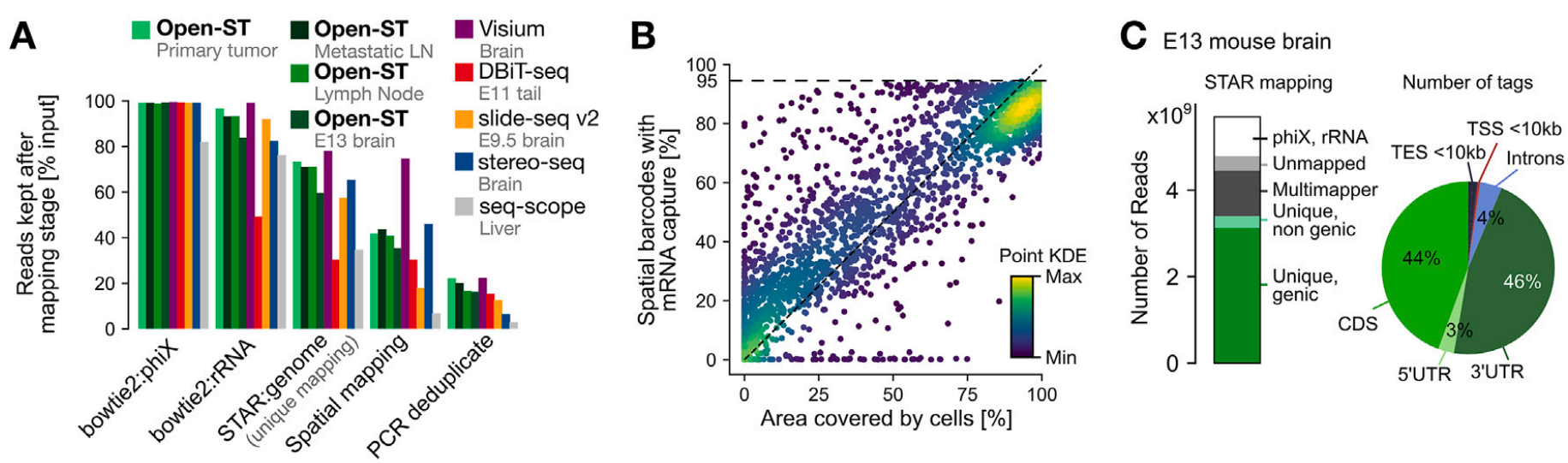
与其他空间转录组技术相比,Open-ST在样本之间始终保持可比或更多的空间映射、去重复读取,Open-ST在处理样本的一致性和精确性上具有优势。转录组文库中检测到的来自第一次测序的条形码读取百分比与组织覆盖率和细胞密度有关,条形码读取的效率受组织结构和细胞分布的影响。图C说明了比对后代表性样本的总读取数的细分。大部分读取被指定为编码序列(CDS)或30个UTR,较少被指定为内含子、基因10kb范围内的转录终止和起始位点(TES/TSS)以及50个UTR,这个细分展示了不同类型的基因区域在数据中的分布情况。
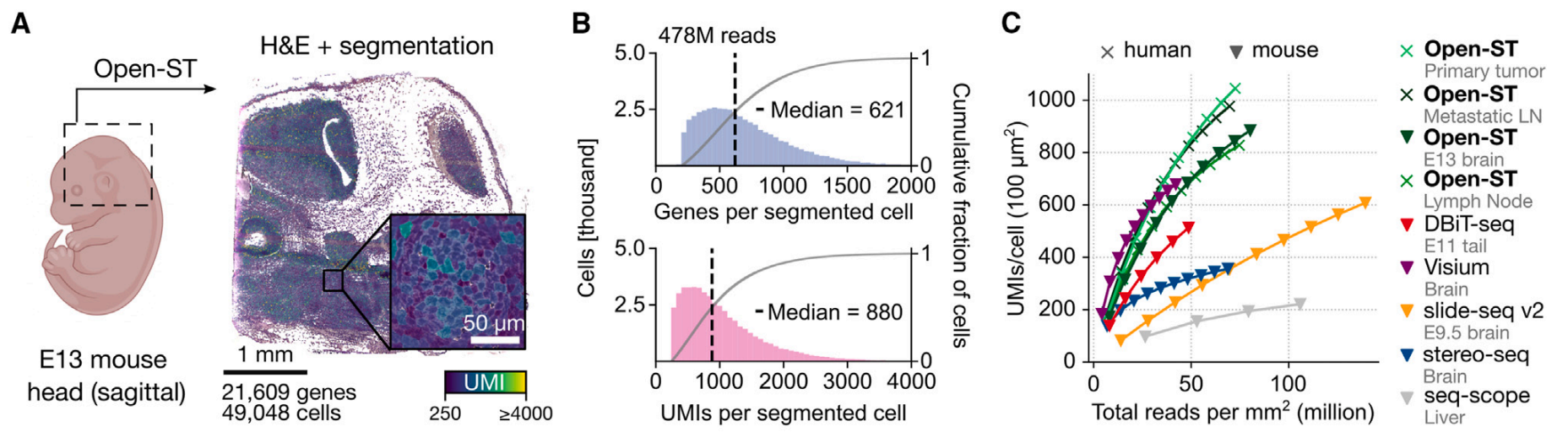
在E13小鼠脑部矢状切片样本中,(A)在它H&E图像中分割出58,881个细胞。测序数据得到49,048个(83%)高质量细胞(R250UMI和<10%线粒体计数),捕获了21,609个基因。
(B)Open-ST高效的RNA捕获产生了中位数为621个基因和880个UMI,其中42%的高质量细胞包含超过1,000个转录本。在所有捕获的转录本中,82% 位于分割细胞内。
(C)与其他ST方法相比,Open-ST 在相同数量的测序读段下,每个细胞的转录本捕获率最高,提高了数据的质量和解析度。
3.Open-ST生成3D虚拟组织块效果展示
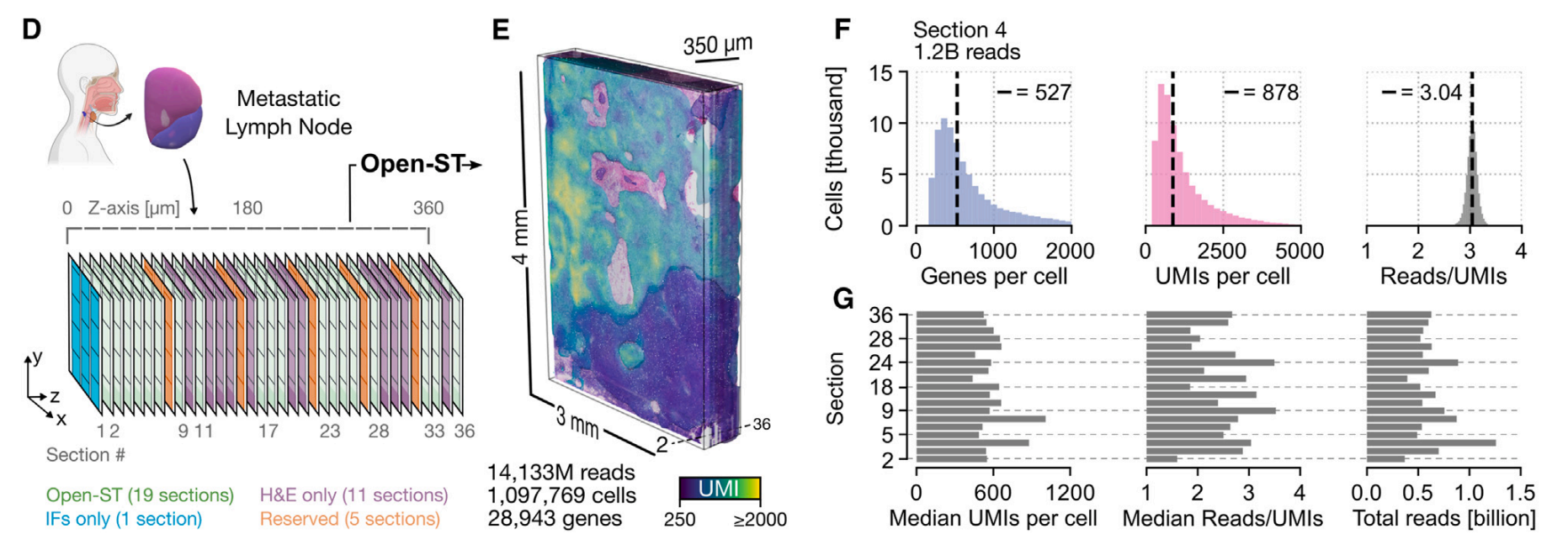
在人类转移性淋巴结样本中,(D)研究获取了10微米的切片,横跨350微米的组织深度,切片分为Open-ST测序用的19张、仅H&E染色的11张、免疫荧光的1张和不做处理的5张分析切片。
(E)在Open-ST测序切片中共获得了超100万个细胞的基因表达谱。用STIM和可视化工具包,将Open-ST处理的连续切片比对,以重建和可视化3D虚拟组织块。空间映射的UMI计数的平滑等值面渲染表明捕获效率可重复,肿瘤区室中可见转录本富集。
(F)第4张切片进行了深度测序(1.2B 读段),显示低读段/UMI (中位数 = 3.04),每个分割细胞的中位数为527个基因,878个UMI。
(G)每平方100mm的读取与UMI比率始终较低,并且在19个切片中相当,除了一个异常值外,证明了 Open-ST 的技术可重复性和成本效益。
Open-ST 可以稳健地捕获连续切片中的转录本,使其非常适合高通量研究和组织的 3D 转录组重建。
4.Open-ST可高精确度局部捕获标记基因,反映核质结构
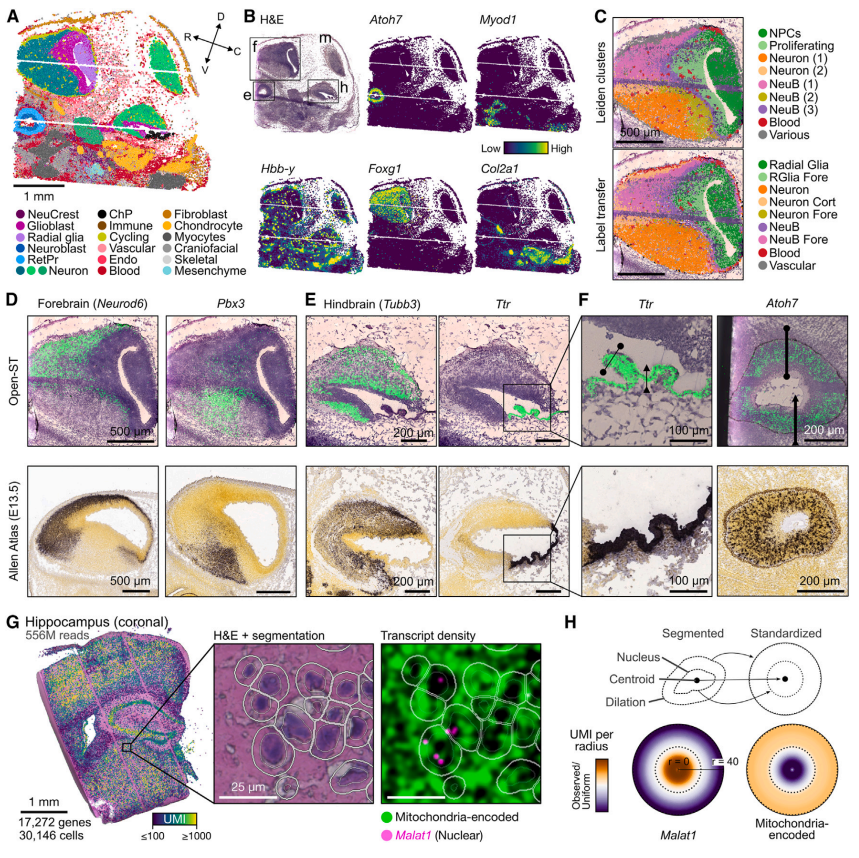
(A-C)图是E13小鼠脑部切片样本测序的聚类情况,检测出的细胞biomarker表达水平良好,说明测序质量不错,能够进行下游分析。
(D-F)图上半部分是Open-ST测序结果,下半部分是 Allen Atlas原位杂交图像,进行Neurod6、Pbx3、Tubb3、Ttr和Atoh7的表达值对比。表明Open-ST局部捕获的基因表达值精确度很高。
(G)左图:每个分割细胞的UMI空间分布反映了组织形态,说明了不同区域的转录活性差异;中图:H&E 分割特写,展现出细胞分割/细胞核分割效果;右图:Malat1主要分布在细胞核中,而线粒体编码的转录本分布在细胞质中,说明了Open-ST技术可以在单细胞水平上检测不同转录本的空间分布,提供了基因表达的空间信息。
(H)图展现了Malat1和线粒体编码转录本在细胞内的具体空间分布情况。说明不同类型的转录本在细胞内具有特定的空间分布特征,通过标准化细胞的可视化方法,可以直观地展示这些分布特征,并进行定量分析。
5.Open-ST可准确剖析原代人体组织中的细胞类型复杂性
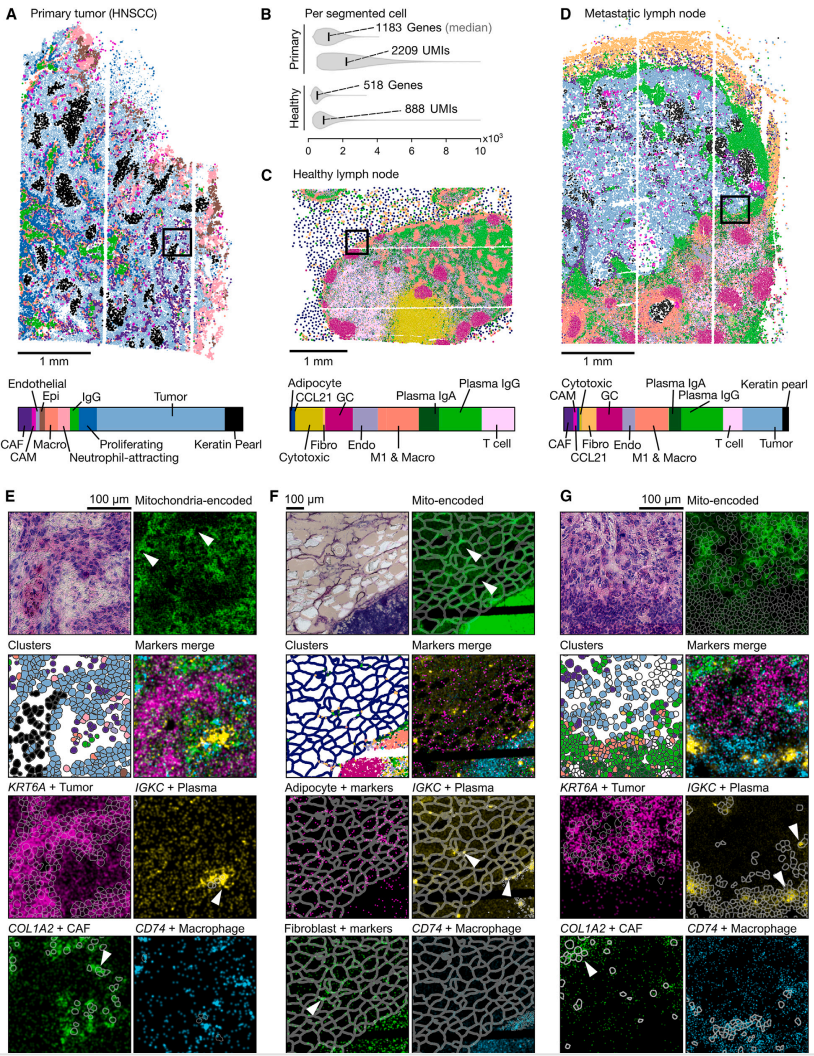
(A-D)图展现了HNSCC、健康淋巴结和转移性淋巴结切片中细胞类型空间分布和细胞类型比例。
(E-G)HNSCC(E)、健康淋巴结(F)和转移性淋巴结(G)中选定的标记基因在空间中的表达。左上:H&E 染色;右上:线粒体编码转录本的表达;第二行:按Cluster分类的分割mask(左)和每个Cluster中标记的基因表达(右);第三行和第四行:不同簇的选定标记的基因表达,以及相应的分割mask。
6.转录组肿瘤异质性在空间分布中具有不同通信特征
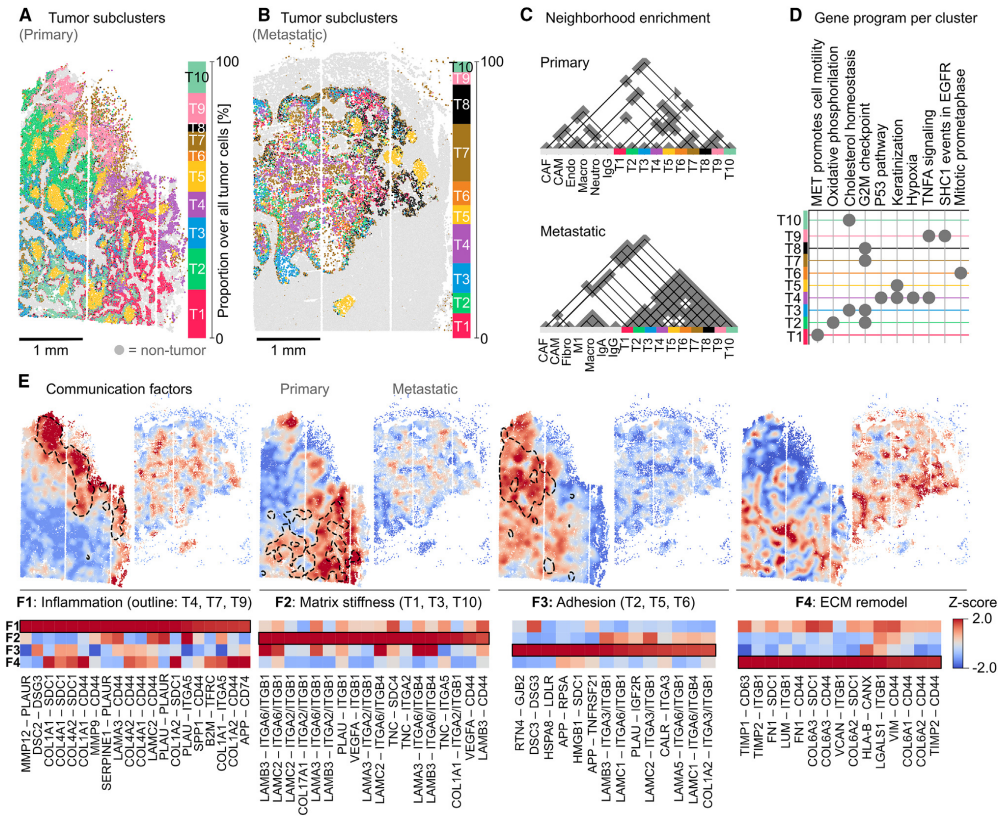
(A-B) 肿瘤细胞在原发性和转移性肿瘤组织中的空间分布和转录组异质性。
(C)肿瘤和基质细胞亚群的空间邻域富集,展现出具有显着空间相互作用的细胞对。
(D)原发性肿瘤组织肿瘤细胞簇中的基因程序归一化富集评分(NES),对不同肿瘤细胞簇进行功能注释。
(E)把肿瘤细胞簇分为四种特征类型,炎症、基质刚度、粘附力和细胞外基质重塑。并展现每个特征类型通信因子评分的空间分布,和每个类型最具代表性的配体-受体对的通信评分。
7.Open-ST 与基于成像的空间转录组学的比较
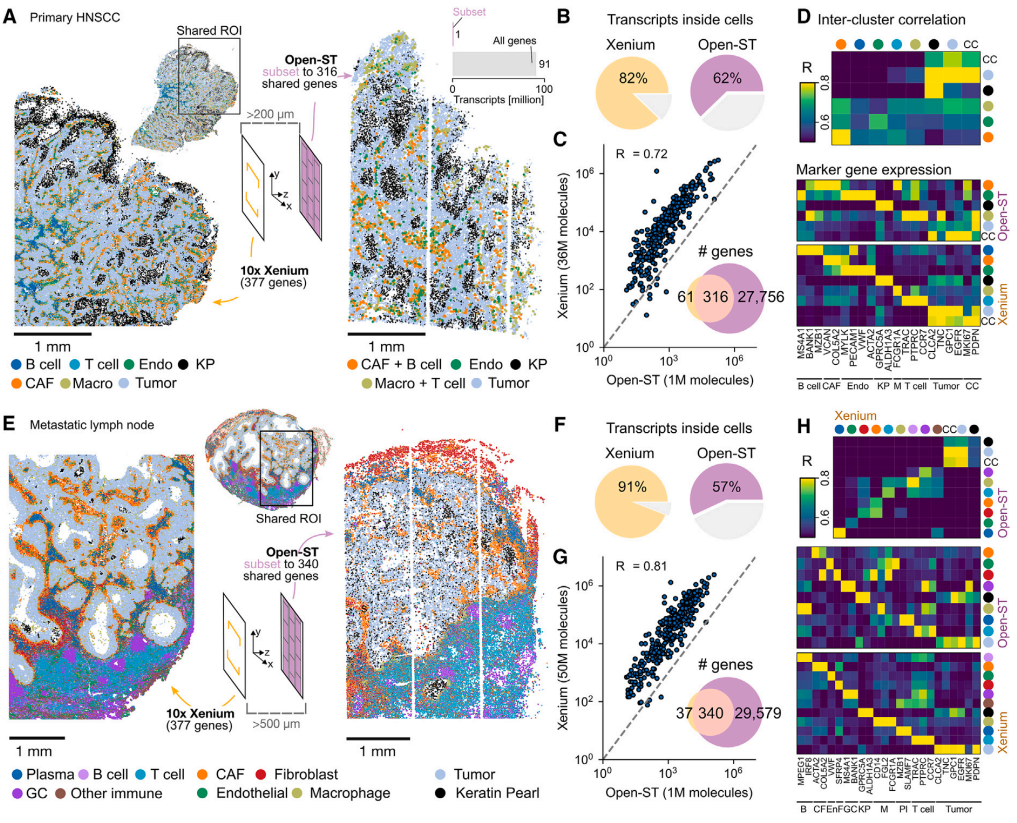
(A)左图:使用Xenium原位平台分析的人类原发性HNSCC组织切片,显示了不同细胞类型的空间分布(如B细胞、T细胞、内皮细胞等)。右图:使用Open-ST平台分析的相同组织切片,显示了与Xenium共享的基因子集(316/377)的空间分布。
(B)转录本比例:Xenium:82%的转录本落在细胞内,Open-ST:62%的转录本落在细胞内。
(C)基因表达相关性:显示了Xenium和Open-ST之间重叠基因的基因表达相关性。相关系数(R):0.72,表示两种技术之间有较高的相关性。
(D)顶部:计算了Xenium和Open-ST中所有inter-cluster的平均归一化基因表达的皮尔逊相关系数。底部:显示了在Xenium和Open-ST数据中原发性HNSCC的细胞亚群标记基因平均表达,按基因归一化。
(E-H)同理。
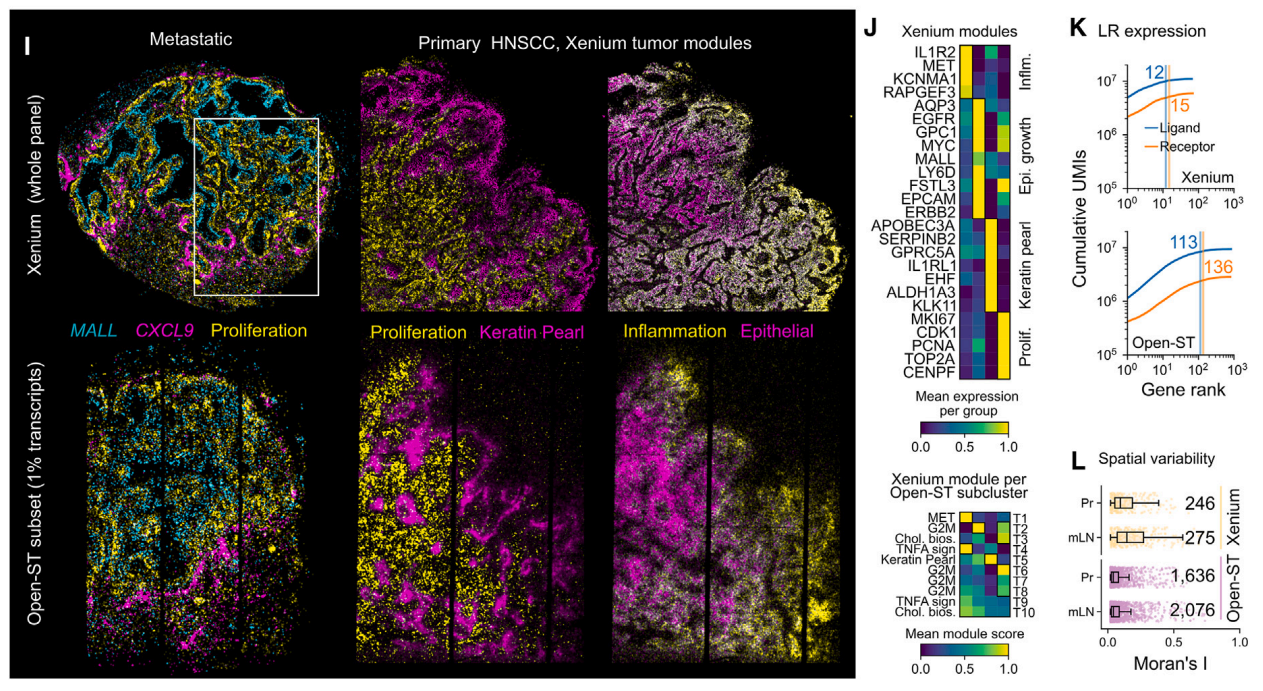
(I)Xenium(上)和Open-ST(下)在原发性HNSCC切片(右)和转移性淋巴结(左)中的基因表达(MALL 和 CXCL9)、增殖相关基因、角蛋白珠相关基因、上皮相关基因和炎症相关基因的空间分布。
(J) Xenium原发组织中每个肿瘤亚簇的基因表达(顶部)和Open-ST 数据(包含 >20,000 个基因)(底部)的每个肿瘤亚群的平均 Xenium 模块得分(模块基因的总体标准化表达)。
(K) 来自 liana 共识数据库的配体-受体基因表达,针对 Xenium(顶部)和 Open-ST 数据(底部)。
(L) Xenium 和 Open-ST 中的空间可变基因。
8. 3D 虚拟组织块:转移性淋巴结

(A) 基因表达的3D可视化:(左)展示了每个细胞中呈现的标记基因的表达水平,包括生发中心的CXCL13、IgG浆细胞的IGHG1、巨噬细胞的LYZ、上皮细胞的KRT6A、CAM的SPP1和CAF的ACTA2。(中)展示了基因表达的平滑处理结果,使得表达水平更加连续和均匀。(右)展示了通过3D渲染生成的平滑表面,显示了基因表达的空间簇。顶部图像展示了免疫区(生发中心、IgG浆细胞和巨噬细胞),底部图像展示了肿瘤和基质区(上皮细胞、CAM和CAF)。
(B)基因表达的3D探索:上图:通过体积剪辑(盒子和自由形式)和平面剪辑技术,展示了ACKR1(小静脉内皮标记)的基因表达在3D虚拟块中的连续性。这种渲染技术允许在不透明的3D虚拟块中以3D形式探索基因表达。下图:平面剪辑显示了基因表达沿z轴的分布,提供了更详细的表达层次信息。
(C)基因表达和组织结构的连续性:(左)展示了H&E组织块的体积剪辑,显示了角蛋白珠结构的连续性。尽管在早期切片中该结构似乎与主要肿瘤块断开连接,但通过体积剪辑可以观察到其连续性。(中)基因表达进一步支持这种连续性,S100A7(角蛋白珠特异基因)和KRT6A(所有肿瘤细胞)在透明组织块中显示了沿z轴的连续表达。(右)由SPP1(CAM)和ACTA2(CAF)表达划定的基质也显示出这种肿瘤结构穿透淋巴结的连续性,边界富含LYZ。虚线表示从H&E确定的肿瘤/免疫边界。
(D) 胆固醇生物合成途径的3D渲染:(左)展示了胆固醇生物合成途径的3D渲染。图中a和b部分展示了两个不同区域的详细视图。(右)放大显示了这些区域中基因表达的分布情况。颜色表示基因表达水平,绿色表示胆固醇生物合成相关基因,黄色表示高表达区域,粉红色表示LYZ(巨噬细胞标记)。
9. 局限性和未来发展
9.1 局限性:
1.本捕获限制:目前仅限于多聚腺苷酸化转录本,存在3’端捕获偏差,限制了对转录本全长和异构体多样性的空间分析。
2.制备局限性:需要调整以保留整个转录本长度,从而更好地实现异构体多样性的分析。
3.特异性:捕获寡核苷酸需要修改和设计,以捕获特定目标如剪接变体或细菌16S转录本。
4.类型适用性:当前不兼容福尔马林固定石蜡包埋(FFPE)组织,FFPE处理会导致RNA碎片化,影响poly(A)捕获效率。
5.与适用性:需要初始资金投入来生成捕获区域,不适合低通量实验,需要合作或机构集中生成捕获区域以降低成本。
6.分割精度:目前由核染色引导,可能导致细胞转录组交叉污染,影响分割精度。
7.与RNA质量:免疫荧光或免疫组织化学染色可能对RNA质量和捕获定位产生负面影响,需要单独优化。
8.异:z轴分辨率(10 µm)与x-y轴分辨率(0.6 µm)的差异可能导致双阳性信号和单细胞水平分析的混淆。
9.分析流程:缺乏标准化的数据分析流程,无法有效解释基于测序的ST方法中的局部偏差、各向异性横向扩散和空间自相关的背景信号。
9.2 未来发展方向
1.本捕获改进:调整文库制备方法以保留整个转录本长度,实现异构体多样性的空间分析。修改捕获寡核苷酸设计,以捕获特定目标如剪接变体或细菌16S转录本。
2.PE兼容性:预计将与FFPE组织兼容,适用于长期储存和更好地保存组织形态。需要额外的脱蜡和解交联步骤以释放RNA,并可能采用基于探针的方法(如Visium FFPE)或原位多聚腺苷酸化技术以捕获碎片化和非多聚腺苷酸化的RNA。
3.成本:通过合作或机构集中生成捕获区域以降低初始资金投入,使得Open-ST适用于低通量实验。
4.分割改进:引入膜染色以提高分割精度,检测多核细胞。优化免疫荧光或免疫组织化学染色条件,以保证RNA质量和捕获定位。
5.分辨率:发展计算方法以反卷积z轴数据,减少分辨率差异对单细胞水平分析的影响。
6.化数据分析流程:发展空间感知的标准化、降维和聚类方法,以解决空间自相关对分析的影响,改进差异表达分析、细胞分型、伪时间和RNA速度分析。

















 1040
1040

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











