









–https://doi.org/10.1101/2025.02.20.639398
研究团队/单位
Vevo Therapeutics; Arc Institute


研究简介
研究意义
构建细胞行为的表达性计算机模型需要生成大型定量数据集,系统地绘制细胞状态如何被各种干扰重塑了构建细胞行为的预测性计算模型。扰动测量能够阐明因果基因-基因相互作用,揭示了反馈回路,并暴露了补偿途径,从而揭示了控制细胞行为和功能的潜在调控网络。
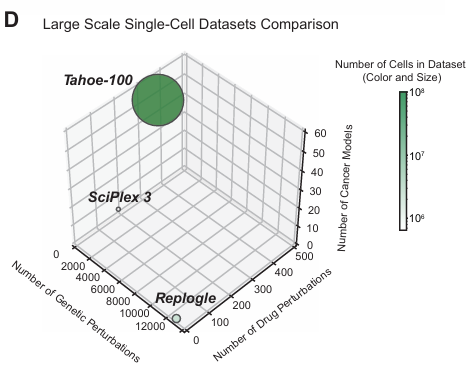
但目前缺乏大规模高通量的基因扰动单细胞测序数据,作为相关计算模型的输入,因此该研究聚焦在这个痛点,发布了1亿规模的单细胞转录组扰动测序数据。
方法和结果
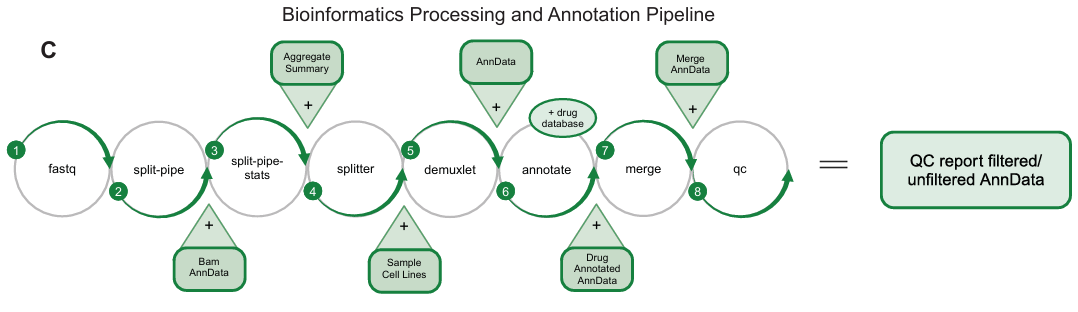
开发 Mosaic 平台,利用单细胞 RNA 测序 (scRNA-seq) 大规模并行捕获扰动下的细胞转录组反应。生成了包含 1 亿+ 单细胞转录组的 Tahoe-100M 数据集,涵盖 50 个癌细胞系和 1,100+ 种小分子扰动。并公开 Tahoe-100M,促进开发细胞行为的 AI 模型,加深对基因功能和调控网络的理解。
Mosaic 平台通过“细胞村”和遗传多重分离,有效减少批次效应,实现大规模扰动研究。
结果
1.用 Vevo 的 Mosaic 平台生成 Tahoe-100M 图谱
研究用 Mosaic 平台测序了 50 个癌细胞系(可以商业购买到的)在加入药物治疗干预后的转录组:
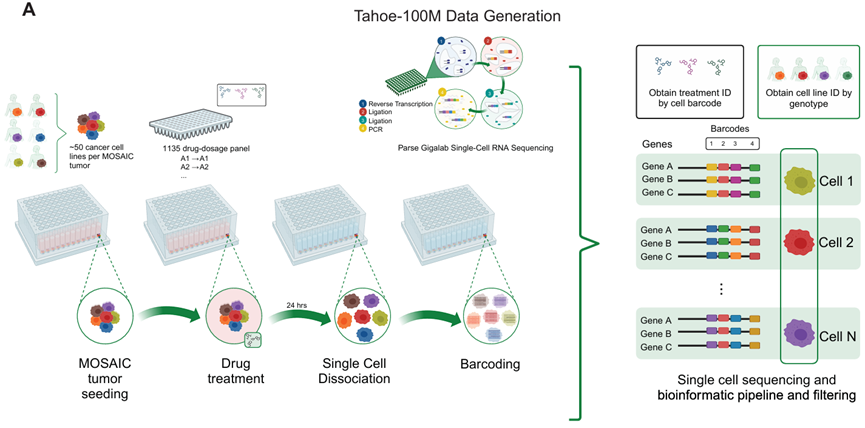
将细胞系混合物组成的球状体悬浮培养,并使每个球状体接受单独的药物治疗,包括 DMSO 载体对照。
药物治疗 24 小时后,球状体被解离、固定,并使用 Parse GigaLab 试剂盒进行分析(一种通过组合条形码实现的、可扩展的 scRNA-seq 分析)。
除了生成不同治疗的单细胞基因表达矩阵外,还检测了 scRNA-seq 文库中存在的遗传变异,并用基于 SNP 的解卷积,将细胞分配到其来源细胞系:
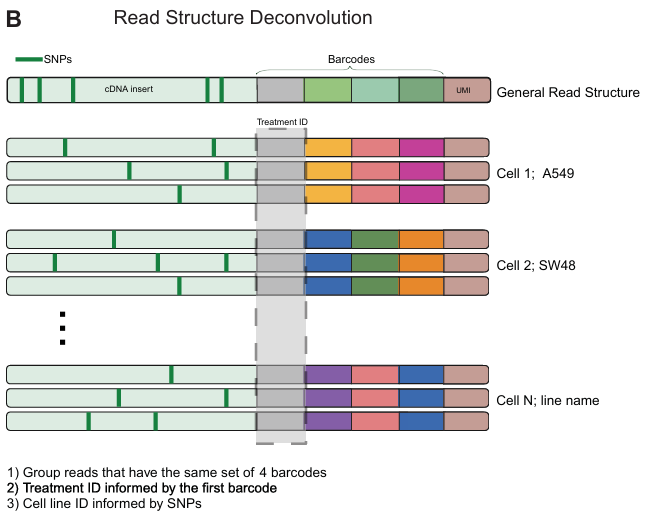
测序得到约 1.4 万亿个原始测序读数,代表总共 1.53 亿个细胞(每个板 820 万到 1450 万个细胞)。
细胞的平均转录本为 2,288 个(中位数为 1,890 个),根据测序深度,每个亚文库的水平存在一些差异。
在使用 Vevo 的过滤标准进行处理后,获得 1.006 亿个通过最小过滤的细胞和 9560 万个通过完整过滤的细胞。研究使用后完整过滤细胞进行此处展示的所有下游分析。
1.1 完整过滤(严格):
- 要求细胞至少具有 700 个独特分子标识符 (UMI)
- 线粒体读数少于 20%
- UMI z 分数在 ±3 以内以去除总计数中的异常值
- 线粒体百分比 z 分数在 ±3 以内以去除线粒体含量中的异常值
- 检测到至少 250 个基因
- 仅保留由 demuxlet 分类为单细胞的细胞,从而排除被识别为双细胞或模糊的细胞
1.2 最小过滤:
- demuxlet 分类为单细胞的细胞(已去除双细胞或模糊的细胞)
- 线粒体读数少于 20%
2.Tahoe-100M 中的细胞系和治疗干预的多样性
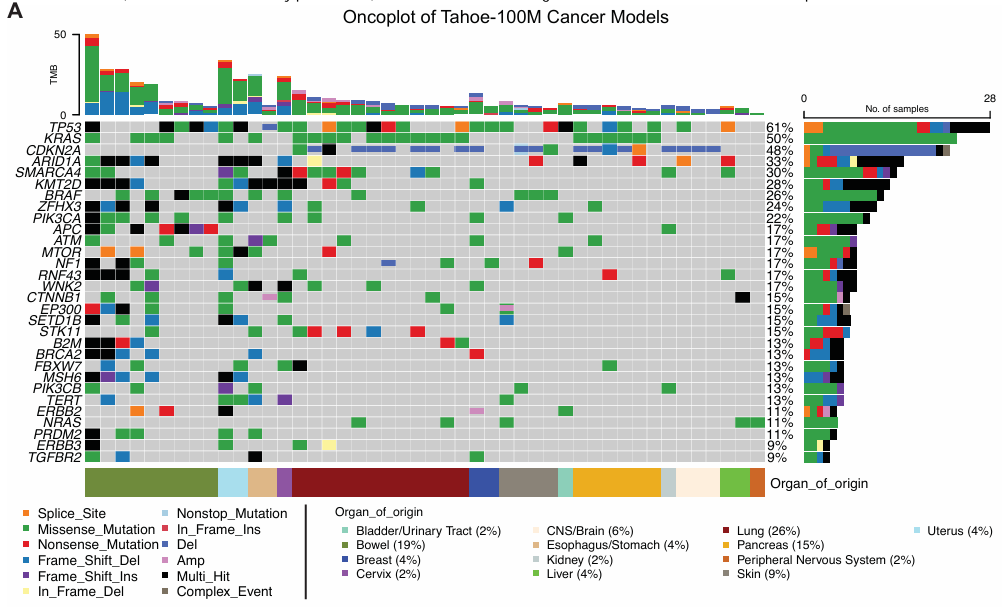
Tahoe-100M 数据集中 47 个细胞系具有足够的代表性,可用于下游分析。47 个细胞系来自 13 个不同的器官,主要包括肺、肠、胰腺和皮肤。细胞系携带多种驱动突变,其中 TP53、KRAS 和 CDKN2A 在约一半的细胞系中发生突变。
2.1 药物特征
-
药物种类:数据集使用了 379 种不同的药物。其中 180 种药物被归类为 25 种作用机制 (MOA),每种 MOA 包含 3 到 27 种不同的药物。大多数(69%)药物为已批准药物,针对多种癌症相关通路。
-
靶点多样性:这些药物靶向 325 个基因,其中 120 个基因被多种药物靶向。
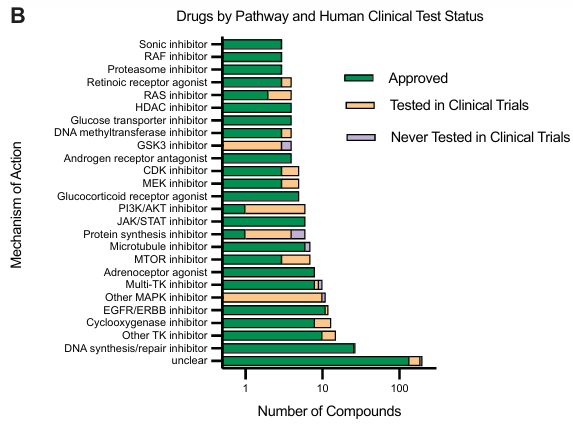
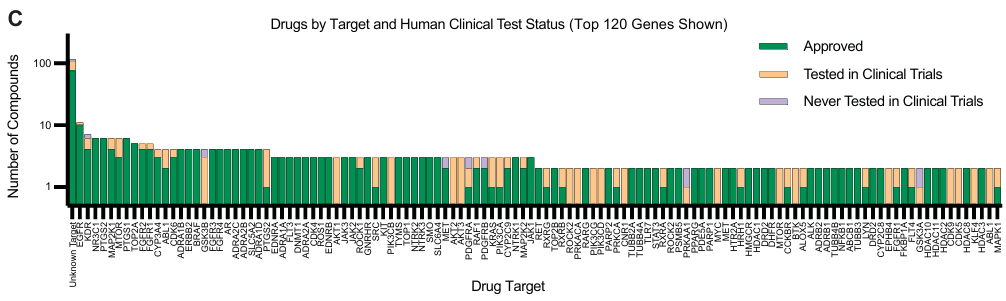
- 数据量:数据集捕获了 17,813 个独特的细胞系-药物条件。与现有基准数据集相比,药物扰动条件数量增加了 31 倍,每个条件的细胞观察数量增加了 29 倍。
3.Tahoe-100M 捕获的全局转录组图谱
3.1 使用 scVI 进行降维
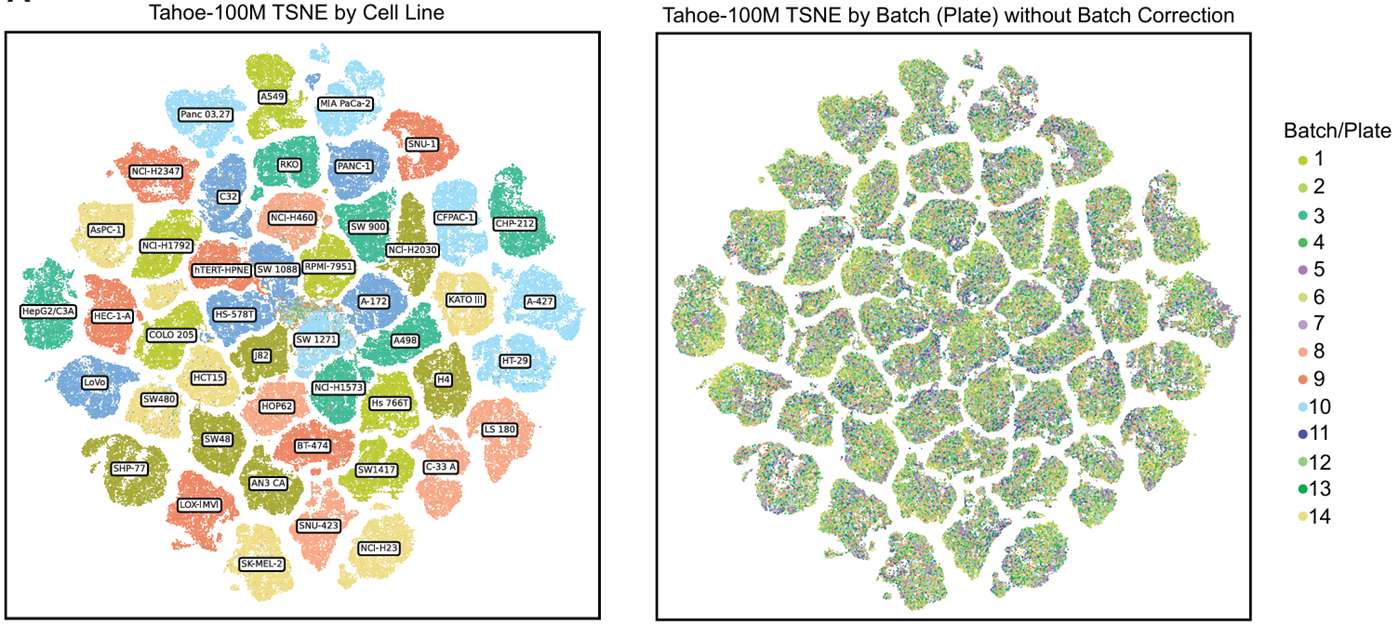
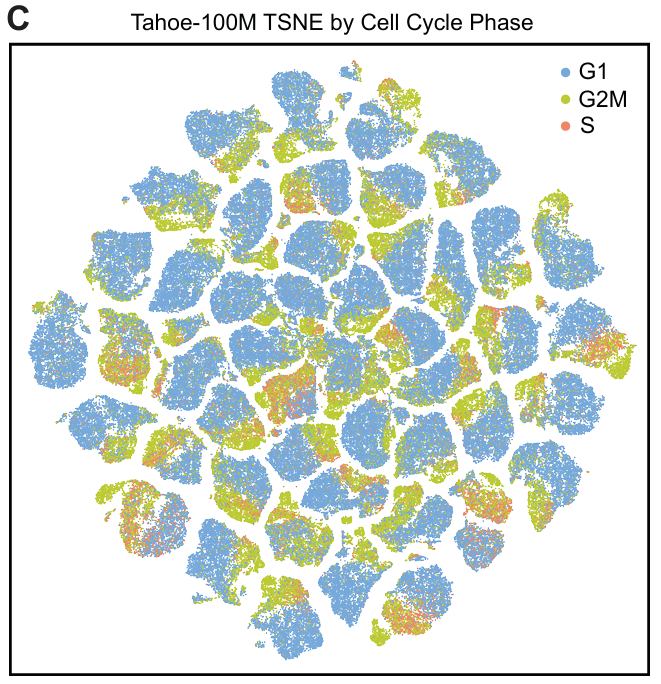
从 47 个细胞系中各采样 140,000 个高质量细胞(总计 658 万个细胞),计算 2-D tSNE 坐标,并绘制 200,000 个随机选择的细胞。tSNE 可视化显示,细胞在转录组空间中基于遗传身份(和细胞周期阶段)而非来源板明显分离,表明数据集中批次效应较小。
3.2 数据集概况
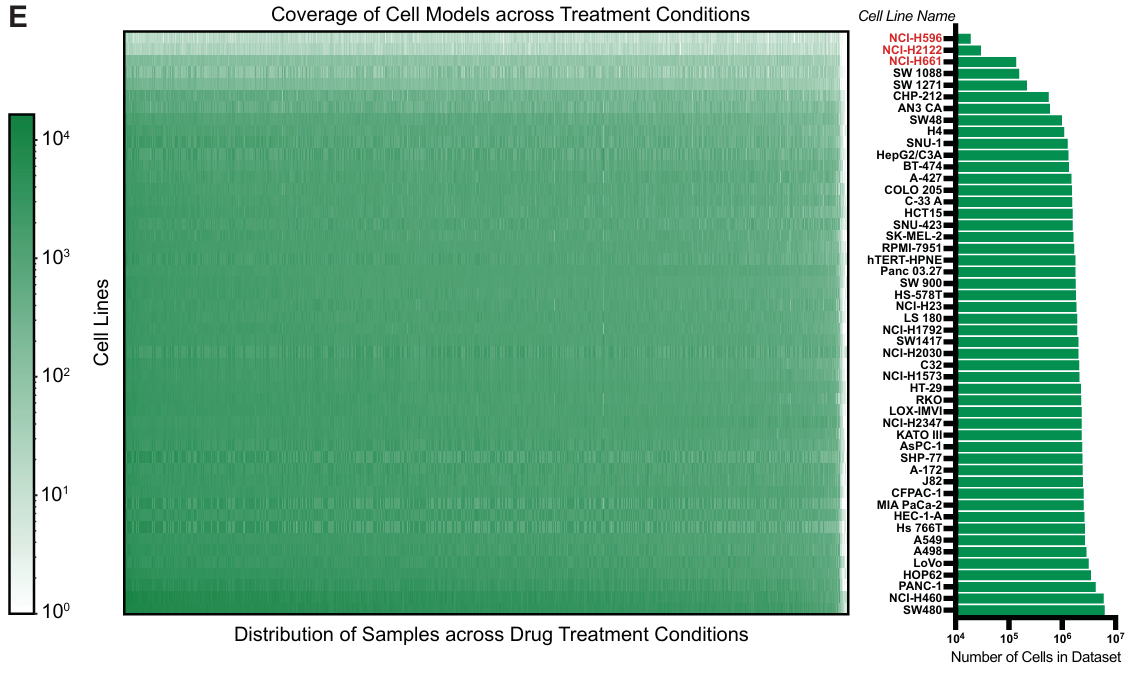
数据集包含 47 个细胞系、379 种药物、1,135 种药物剂量组合和 52,886 个独特的细胞系-药物-剂量条件。每个条件的中位数为 1,287 个细胞。
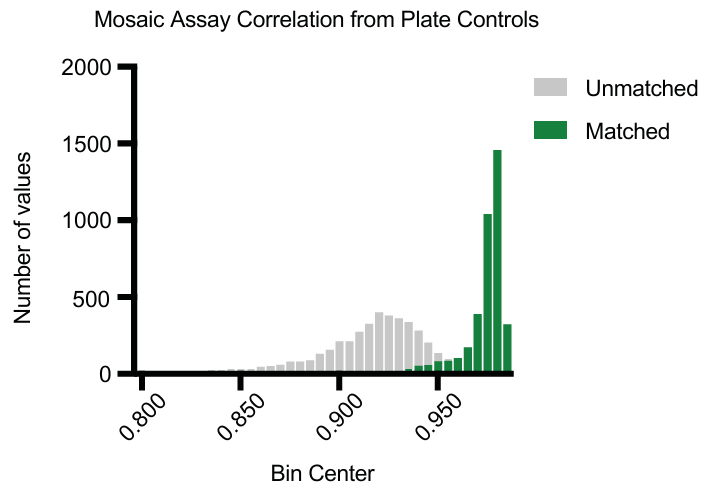
3.3 Mosaic 平台的可重复性
将第 14 板作为第 6 板的生物学重复,评估 Mosaic 平台的可重复性。
对来自这些板的未转换基因表达矩阵进行伪批量分组,并计算 Pearson 相关性。
结果显示,匹配治疗和细胞身份的条件之间的相关性较高(中位数:0.975),而不匹配条件之间的相关性较低(中位数:0.915),表明平台具有良好的可重复性。
4.跨细胞系和作用机制的药物诱导转录组效应
为了深入了解 Tahoe-100M 数据集中药物诱导的转录组变化,研究进行了以下分析:
4.1 影响细胞分组的因素
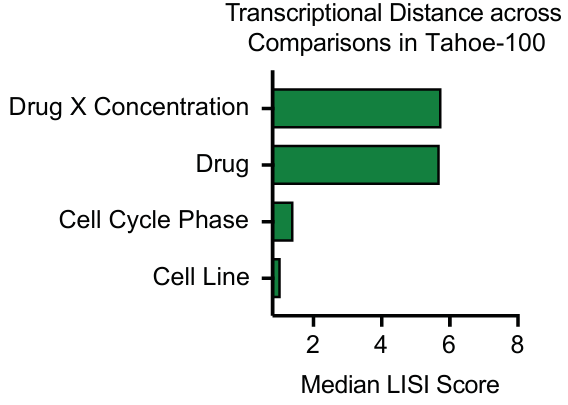
使用局部逆辛普森指数 (LISI) 评估不同元数据因素对细胞分组的影响。
结果显示,细胞系身份和细胞周期阶段是驱动数据分层的最主要因素。
药物治疗和剂量的影响相对较小,需要进行差异基因表达分析才能更准确地描绘药物诱导的变化。
4.2 药物治疗的转录组反应
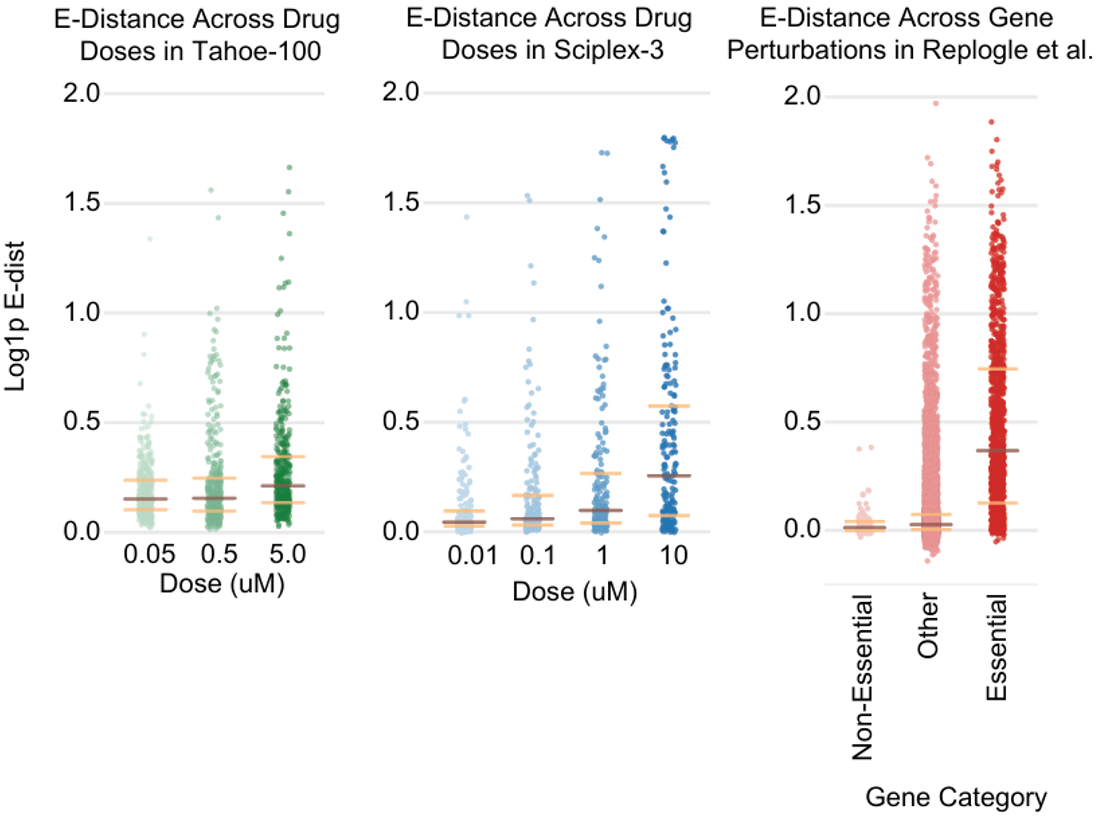
使用 E-distance 研究给定治疗在整个转录组中的效应大小,这个分析在单细胞扰动数据中基于距离度量分析,量化了扰动群体与其对照的可分离性。研究使用 scVI 嵌入计算了细胞系内所有药物条件的 E-distance。
结果观察到最高药物剂量下中位数 E-distance 较大,表明剂量与效应大小呈正相关(图左)。与其他单细胞扰动数据集(Sciplex3(中)、CRISPRi(右))相比,Tahoe-100M 中的药物扰动表现出中等效应。
4.3 不同作用机制 (MOA) 的药物效应
按报告的 MOA 对药物进行分层,发现不同 MOA 的药物效应存在差异。
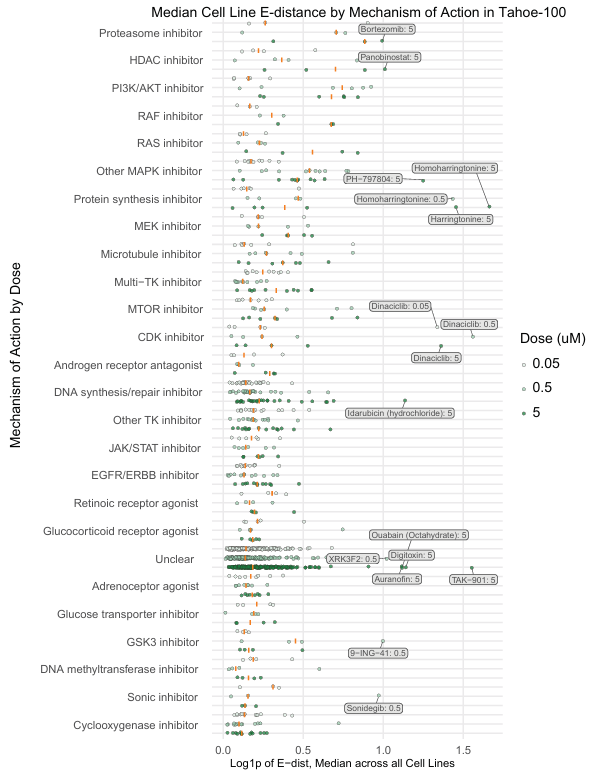
-
哈灵顿碱和高哈灵顿碱、dinaciclib 等药物表现出显著的异常效应。
-
蛋白酶体抑制剂、HDAC 抑制剂和 PI3K/AKT 抑制剂在剂量水平上往往具有更大的效应。
-
作用于上皮实体瘤的抗癌药物(MAPK、PI3K/AKT、MTOR 和 CDK 抑制剂、微管干扰剂和 DNA 合成/修复抑制剂)往往比其他药物具有更大的 E-distance。
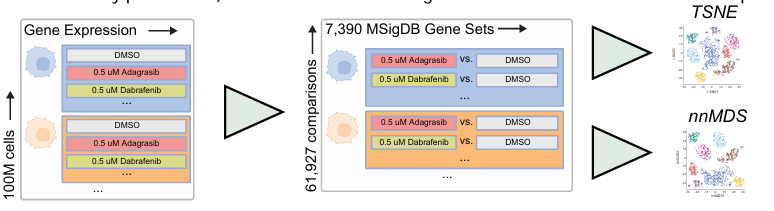
4.4 基于差异表达基因集评分的降维
将细胞系、药物和剂量的每个独特组合视为单个数据点,进行降维分析。
t-SNE 和 nnMDS 可视化显示出清晰的 E-distance 梯度,表明药物效应大小与数据点分布相关。
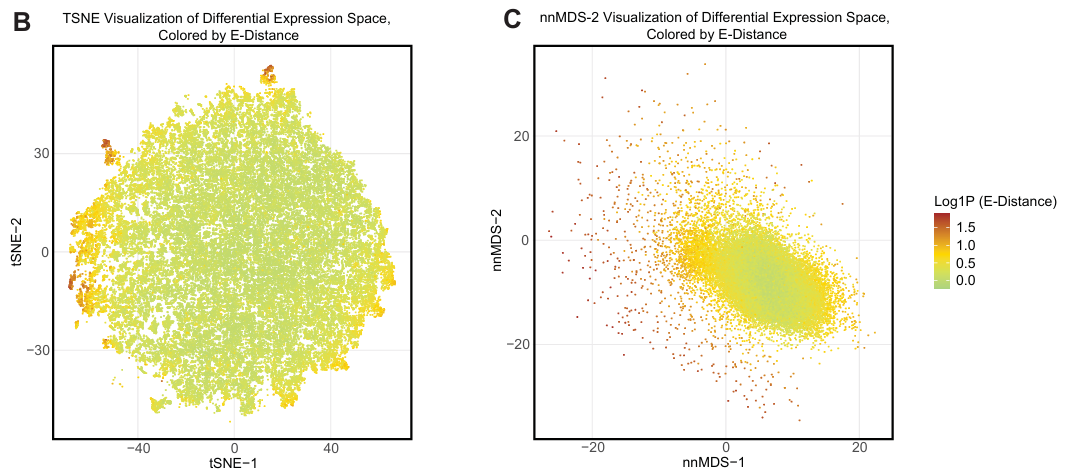
不同 MOA 的药物在可视化空间中呈现重叠但也有分离,尤其是在高 E-distance 区域。
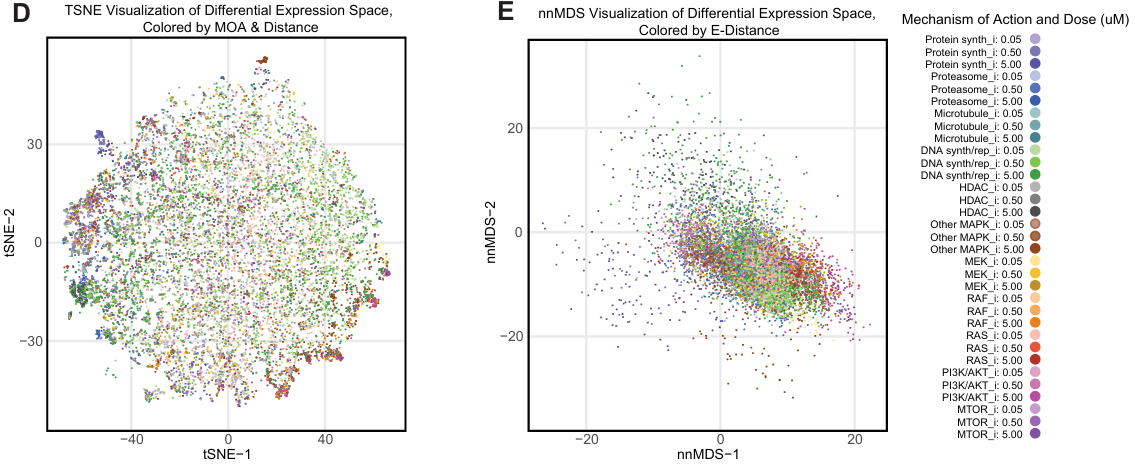
KRAS G12C 特异性抑制剂 Adagrasib 和 Pan-RAS 抑制剂 RMC-6236 在 KRAS G12C 细胞系中显示出不同的分布,符合预期。
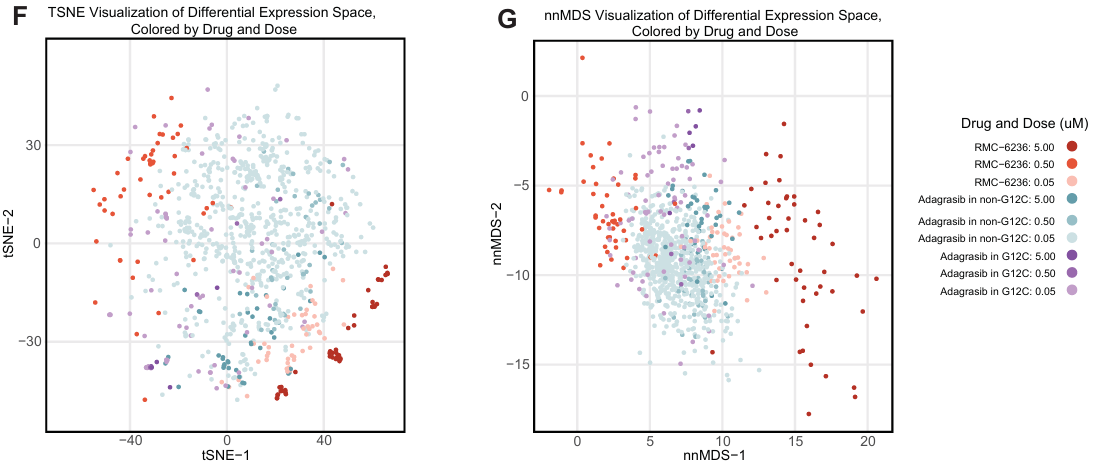
5.RAS/RAF 通路的上下文依赖性抑制
为了深入研究药物的转录效应,研究重点关注了 RAS/RAF 通路的靶向治疗,并分析了药物对细胞周期阶段分布的影响,作为一个分析这批数据的示例。
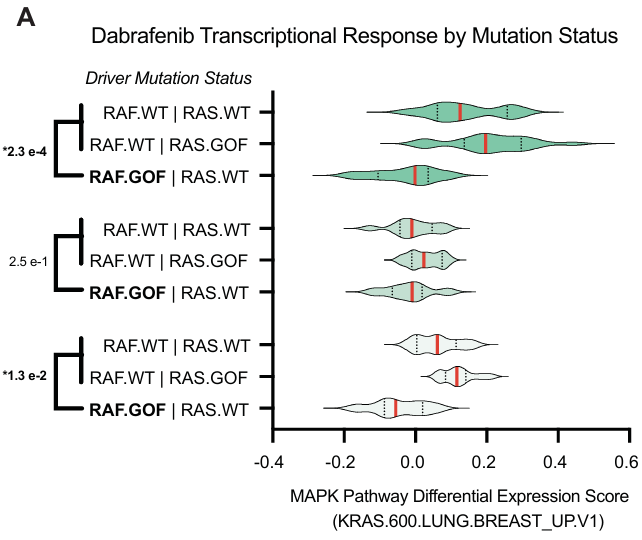
研究使用 Vision 计算细胞水平的基因集表达评分,然后计算聚合治疗和对照条件之间的差异评分。采用了 MSigDB.c6 基因集 ,研究之前将其确定为 RAS/RAF 通路活性的高效转录的特征。
5.1 RAS/RAF 通路抑制剂的效应
Dabrafenib (BRAF-V600E 抑制剂)在三种细胞系分层中该特征的差异 Vision 评分:
在携带 BRAF-V600E 突变的细胞系中,Dabrafenib 显著降低了 RAS/RAF 通路活性的转录特征评分。在携带 KRAS 突变的细胞系中,Dabrafenib 的效应较小。这些结果符合 RAS/RAF 信号通路的预期动态,即 BRAF 突变发生在 KRAS 突变的下游。
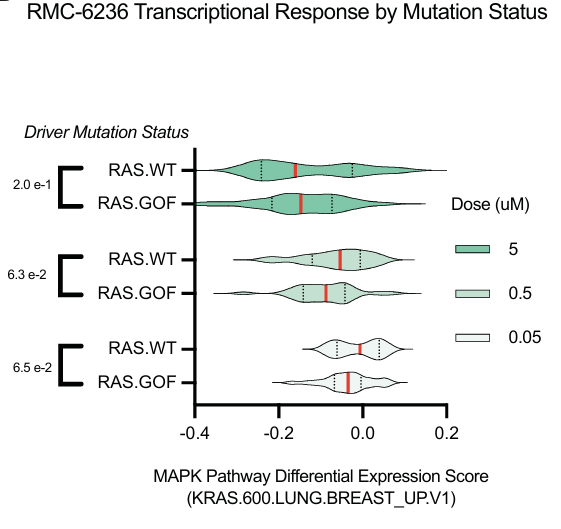
RMC-6236 (pan-RAS 抑制剂) 在 KRAS 突变细胞系中产生了最大的效应,降低了 KRAS 信号。
这些结果强调了 RMC-6236 的 KRAS 特异性作用。药物对细胞周期阶段分布的影响
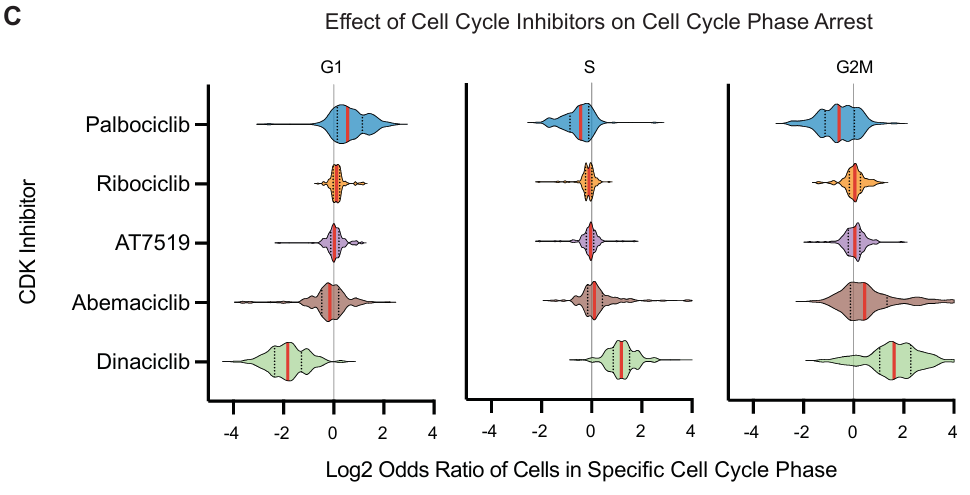
CDK 抑制剂:
- Palbociclib 在 G1 期显示富集,Dinaciclib 在 G2/M 期显示富集,这与其他 CDK 抑制剂的效应不同。
- Dinaciclib 抑制 CDK1、CDK2、CDK5 和 CDK9,诱导 G2/M 期阻滞。
- Palbociclib 选择性靶向 CDK4/6,促进 G1 期到 S 期的转变。
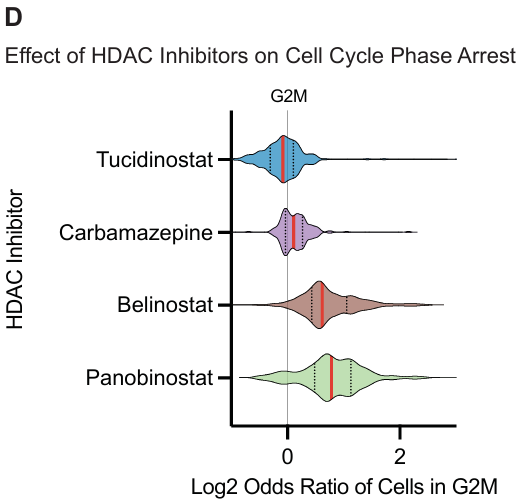
HDAC 抑制剂:
- Belinostat 和 Panobinostat 主要驱动了 G2/M 期富集。
- 卡马西平的富集缺失可能归因于其主要作用是电压门控通道调节剂。
- Tucidinostat 的负富集可能是由于其抑制 HDAC1、HDAC2、HDAC3 和 HDAC10,诱导 G0/G1 细胞周期阻滞。
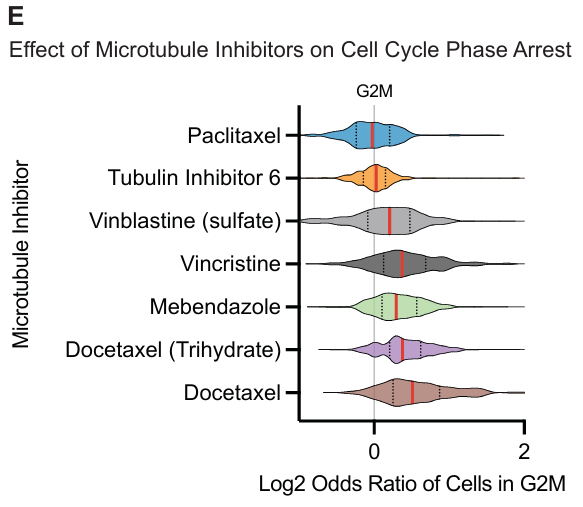
微管抑制剂:
- 大多数微管抑制剂在 G2/M 期产生富集,但长春花碱和微管蛋白抑制剂 6 是例外。
- 微管抑制剂通过激活有丝分裂检查点,诱导有丝分裂阻滞。
未来方向
通过公开发布 Tahoe-100M,作者的目标是播下数据驱动发现和人工智能模型开发的良性循环的种子。研究人员不仅可以使用该资源来改进现有的计算框架,还可以解锁对基础细胞生物学、药物疗效和治疗耐药性的新见解。随着该领域围绕大规模扰动图谱的融合,实验和计算创新之间的协同作用将使我们更接近对细胞的预测性、机制性理解,并最终实现更有效、个性化的患者治疗。
感谢Vevo Therapeutics; Arc Institute的成果贡献!
下载数据
Tahoe-100的数据的详细信息可以在这里获取:
https://github.com/ArcInstitute/arc-virtual-cell-atlas/blob/main/tahoe-100/README.md
作者提供的是最小过滤标准的数据,即1344个样本,合计100648790个细胞。
数据存储在Google Cloud Storage当中,连接是gs://arc-ctc-tahoe100/
我个人使用的下载方法:
1.安装google cloud sdk到你的电脑or服务器上
注意,安装Google SDK时要求安装Python 2.7或以上版本,如果你是conda环境,在用google cloud sdk的时候还需要激活你的conda环境(python版本高一些)。
下载地址:
https://cloud.google.com/sdk/docs/install?hl=zh-cn#linux
依照你的需要下载即可
然后就是解压和安装:
tar -xf google-cloud-cli-linux-x86_64.tar.gz
./google-cloud-sdk/install.sh
也可以通过提供偏好设置作为标志以非交互方式(例如使用脚本)完成此操作。如需查看可用标志,请运行以下命令:
./google-cloud-sdk/install.sh --help
如需发送匿名使用情况统计信息以帮助改进 gcloud CLI,请在出现提示时回答 Y。
如需将 gcloud CLI 添加到您的 PATH 并启用命令补全功能,请在出现提示时回答 Y。如果更新了 PATH,记得要打开新终端,使更改生效。
最后,初始化 gcloud CLI:
./google-cloud-sdk/bin/gcloud init
2.查看Tahoe-100M数据和下载
安装完之后就可以cd到google-cloud-sdk的目录下使用它bin文件夹里的下载命令。记得先激活你的conda环境,或你当前python版本高于2.7的环境都可以。
你可以用这里的命令查看它目录下有什么:
gsutil ls gs://arc-ctc-tahoe100/2025-02-25/

你可以继续看下一级目录内容:
gsutil ls gs://arc-ctc-tahoe100/2025-02-25/h5ad/
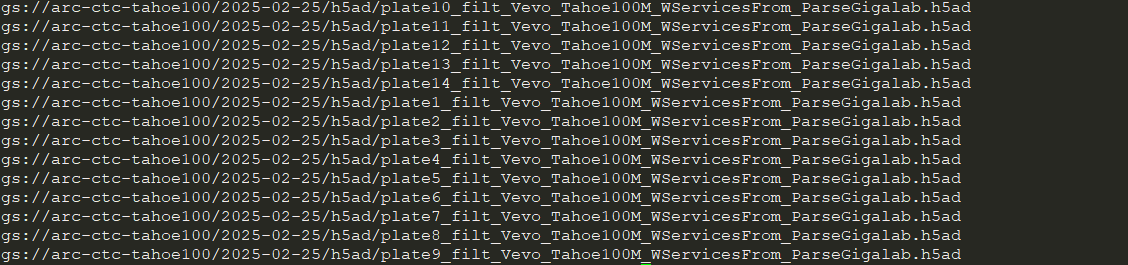
h5ad文件夹就是作者提供的数据,metadata文件夹就是对应的单细胞数据信息了(药物、细胞系…)
把后h5ad文件里的全部数据下载到你的目录中(313个G):
gsutil -m rsync -r gs://arc-ctc-tahoe100/2025-02-25/h5ad/ /你的路径/
metadata一样的操作
作者github上使用的方法:
https://github.com/ArcInstitute/arc-virtual-cell-atlas/blob/main/tahoe-100/tutorial-py.ipynb
1.配置好环境
which conda && conda env create -q -f ../conda_envs/python.yml
2.用python获取每个样本的metadata
import os
import io
import pandas as pd
import scanpy as sc
import pyarrow.dataset as ds
import gcsfs
# 初始化GCS 文件系统以从GCS 读取数据
fs = gcsfs.GCSFileSystem()
# GCS 存储桶路径
gcp_base_path = "gs://arc-ctc-tahoe100/2025-02-25/"
# 样本meta数据的路径
infile = os.path.join(gcp_base_path, 'metadata', 'sample_metadata.parquet')
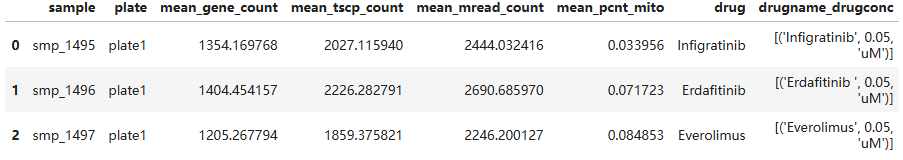
# 只读取前 3 行
sample_metadata = ds.dataset(infile, filesystem=fs, format="parquet").head(3).to_pandas()
sample_metadata
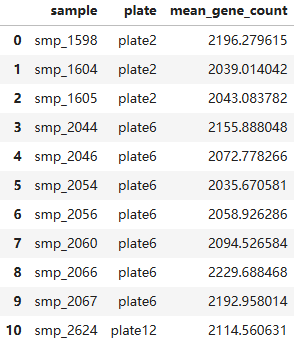
# 选择某些列和行进行过滤
columns_to_read = ['sample', 'plate', 'mean_gene_count']
dataset = ds.dataset(infile, filesystem=fs, format="parquet")
sample_metadata = dataset.to_table(filter=(ds.field('mean_gene_count') > 2000), columns=columns_to_read).to_pandas()
sample_metadata
# 获取样本数量(1344)
columns_to_read = ["sample"] # Specify the columns you need
dataset = ds.dataset(infile, filesystem=fs, format="parquet")
sample_count = dataset.to_table(columns=columns_to_read).to_pandas()["sample"].nunique()
print(f"Number of samples: {sample_count}")
# 每plate的样本数目
columns_to_read = ["plate", "sample"] # Specify the columns you need
dataset = ds.dataset(infile, filesystem=fs, format="parquet")
samples_per_plate = dataset.to_table(columns=columns_to_read).to_pandas().groupby("plate").size()
samples_per_plate
3.用python获取每个细胞的metadata
# 设置 obs_metadata 文件的路径
infile = os.path.join(gcp_base_path, 'metadata', 'obs_metadata.parquet')
# 读取meta数据的子集
obs_metadata = ds.dataset(infile, filesystem=fs, format="parquet").head(100000).to_pandas()
obs_metadata
只展示前10000个
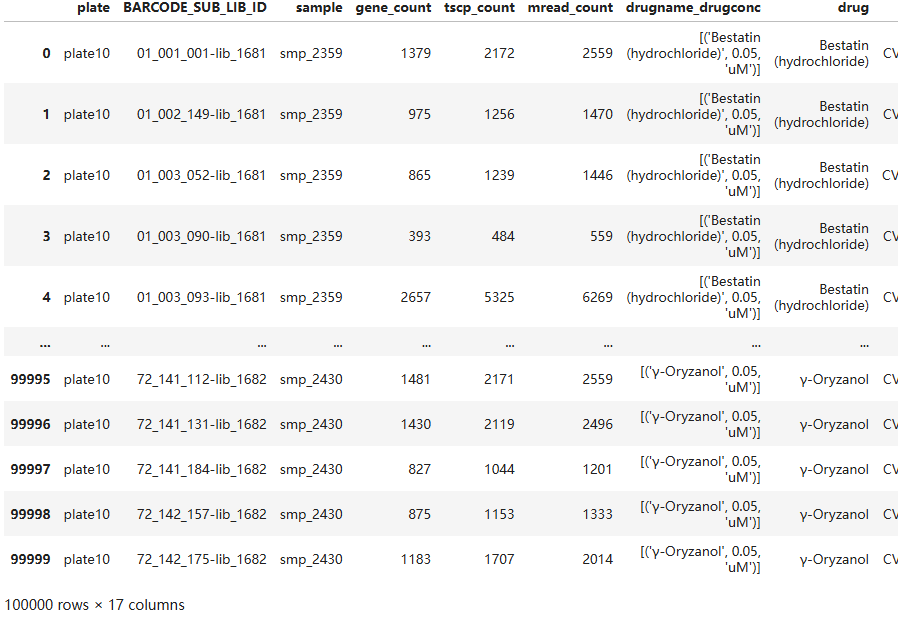
样本数量
obs_metadata["sample"].nunique()
基因计数分布
pd.options.display.float_format = '{:.0f}'.format
obs_metadata["gene_count"].describe()
tscp(UMI)计数分布
pd.options.display.float_format = '{:.0f}'.format
obs_metadata["tscp_count"].describe()
4.读取h5ad文件
对于本教程,将读取 1 h5ad 文件的子采样版本,因为每个板的 h5ad 文件都相当大。
infile = "gs://arc-ctc-tahoe100/2025-02-25/tutorial/plate3_2k-obs.h5ad"
with fs.open(infile, 'rb') as f:
adata = sc.read_h5ad(f)
adata

查看细胞的信息
print(adata.obs.shape)
adata.obs.head()
5.下载数据
例如:
gsutil cp gs://arc-ctc-tahoe100/2025-02-25/tutorial/plate3_2k-obs.h5ad
大数据的时候
gsutil rsync gs://arc-ctc-tahoe100/2025-02-25/tutorial/ /你的路径/




















 416
416

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











