







相对于PyMol来说,ChimeraX可能扩产性差一点(毕竟PyMol是原生Python,我写起辅助脚本来比较简单,而且Pymol给了很好的开源文档)。但是Chimera-X的预设实在是过于强大了,但是如果利用好Chimera的预设风格,很简单就能画出PyMol很难制作的图片
一,chimerax的安装
1,安装chimerax:可以使用pymol平替,参考我的上一篇博客
https://blog.csdn.net/weixin_62528784/article/details/144698291?spm=1001.2014.3001.5501
https://www.cgl.ucsf.edu/chimerax/download.html
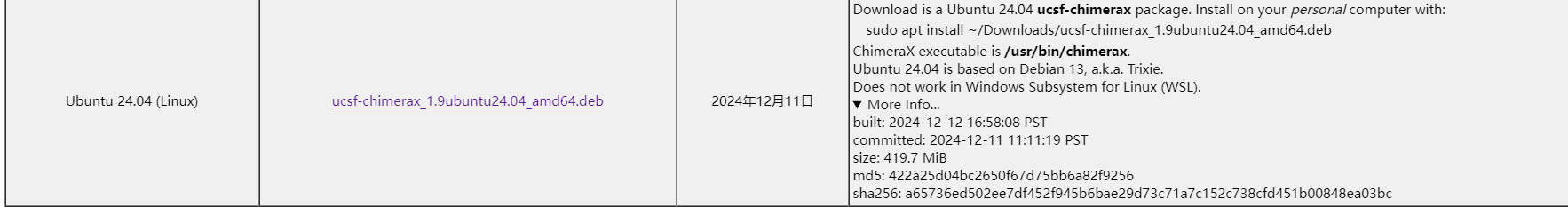
建议使用curl下载,现在使用wget都不太灵了
curl -o ucsf-chimerax_1.9ubuntu24.04_amd64.deb "http://example.com/chimerax-get.py?file=1.9%2Fubuntu-24.04%2Fucsf-chimerax_1.9ubuntu24.04_amd64.deb"

建议带上./,不然会文件会识别失败导致安装失败
sudo apt install ./ucsf-chimerax_1.9ubuntu24.04_amd64.deb
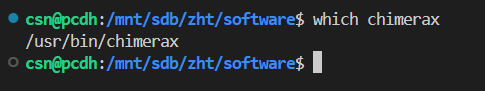
安装好之后的软件就在 /usr/bin/chimerax,所以直接就在环境变量中了,可以直接使用
然后,同理因为chimerax需要图形可视化界面,所以我们还是在x-server的mobaxterm上打开
2,获取目标蛋白质结构数据:
直接从uniprot中获取:
https://www.uniprot.org/uniprotkb/P49711/entry#structure
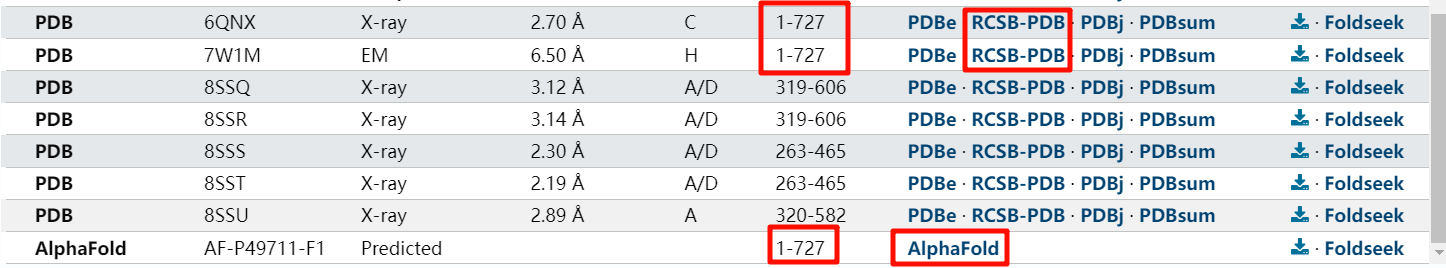
以最后的alphafold的结构数据为主
3,chimerax使用教程:
https://chimera-linux.org/docs/
https://www.cgl.ucsf.edu/chimera/docindex.html
https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/frametut.html
https://dasher.wustl.edu/chem430/software/chimera/users-guide.pdf
建议直接看英文教程,不要看二手中文博客教程,至于学术英语翻译插件已经提供,可以参考我的之前的web博客
二,结构生物学可视化选择语法:以pymol为例
在分子可视化和分析中,PyMOL 是一个广泛使用的工具,它不仅能展示分子的三维结构,还能帮助我们深入分析分子的各种性质。PyMOL 选择器语法是其核心功能之一,它允许用户精确地选择分子中的特定原子、残基或区域,进行各种操作,如颜色设置、距离测量、计算分子间相互作用等。通过灵活运用选择器语法,我们能够高效地从复杂的分子结构中提取出感兴趣的部分,进行定制化的分析与可视化。无论是在药物设计、分子对接研究,还是在结构生物学领域,PyMOL 选择器语法都为研究人员提供了强有力的工具,提升了工作效率,简化了数据分析过程。
看到一篇不错的博客,展示的图如下:
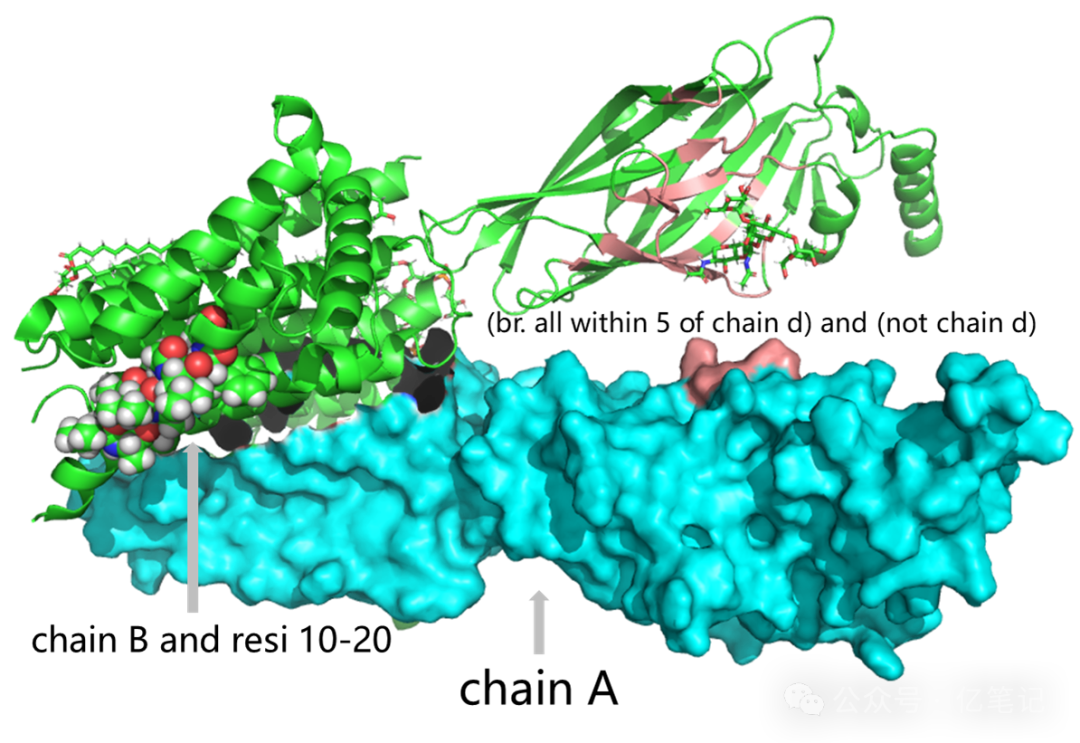
这里加点我个人的私货:
选择是pymol、chimeraX等结构生物学工具的核心,只要掌握了选择语法,其他的都是看文档help、随看随学的难度。
先select,再action
pymol选择器简介及语法
PyMOL的选择语言允许根据标识符和属性选择object中的特定原子。这种方式可以灵活选择和操作目标分子或原子,精确控制可视化和分析过程。许多命令(如 color、show 等)接受原子选择参数,仅对场景中的部分原子操作(改变显示类型,透明度,颜色等),非常方便。此外还可以通过逻辑运算符(如 and、or 和 not)将选择条件变得更精确或更具包容性,and表示选择同时具备所有指定属性的项目。or表示选择具备任意指定属性的项目。
PyMol中可以使用两种方式执行选择器命令。
方式一:如下在图形界面中使用select命令或者cmd.select()命令进行选择。
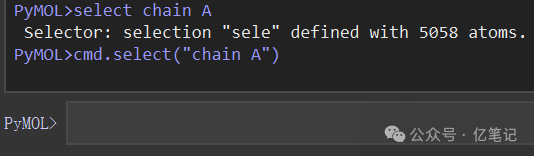
select chain A
cmd.select("chain A")
方式二:可以在脚本中导入cmd,然后使用cmd.select()进行选择。
from pymol import cmd
cmd.select("chain A")
选择操作符及示例
通用选择(Generic)

select all
select none
select enabled
命名的选择操作(Named selections)

select sele
select %sele
select ?sele
逻辑选择运算符(Logical)
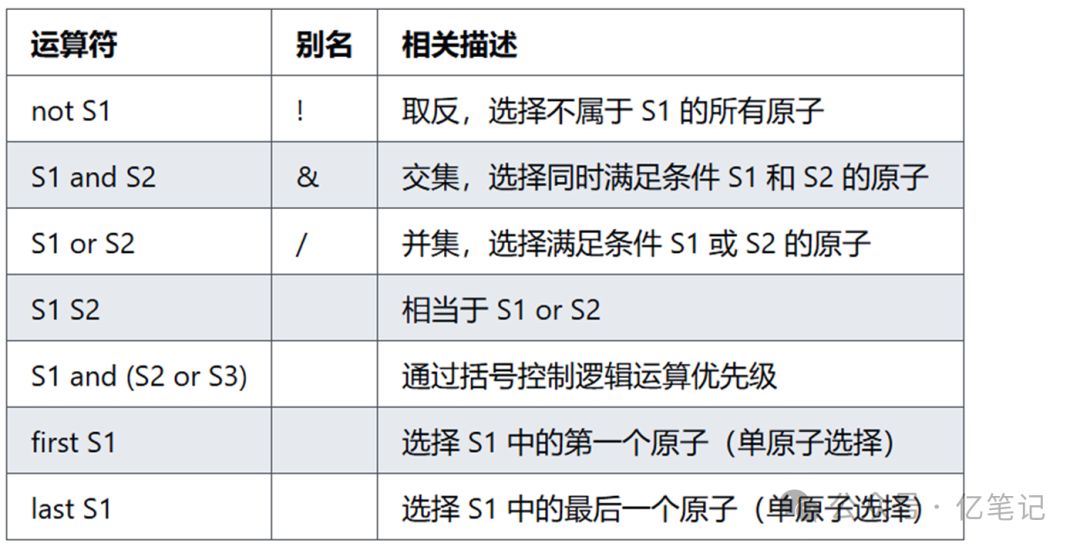
select not chain A # 选择不在链 A 中的所有原子。
select chain A and resi 50 # 选择链 A 中编号为 50 的残基原子。
select chain A or chain B # 选择链 A 或链 B 中的原子。
select chain A chain B # 等效于 chain A or chain B。
select chain A and (resi 50 or resi 100) # 选择链 A 中编号为 50 或 100 的残基原子。
select first chain A # 选择链 A 中的第一个原子。
select last chain A # 选择链 A 中的最后一个原子。
标识符(Identifiers)
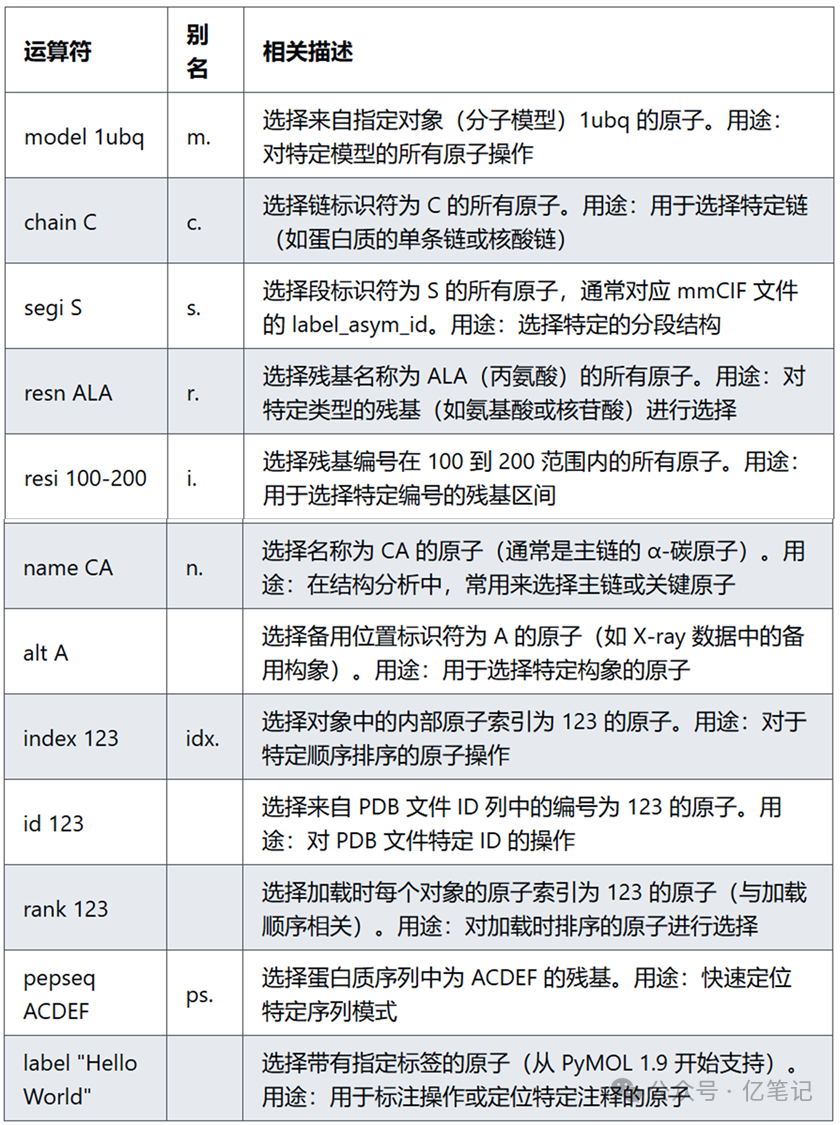
select model 1ubq
select chain C
select segi S
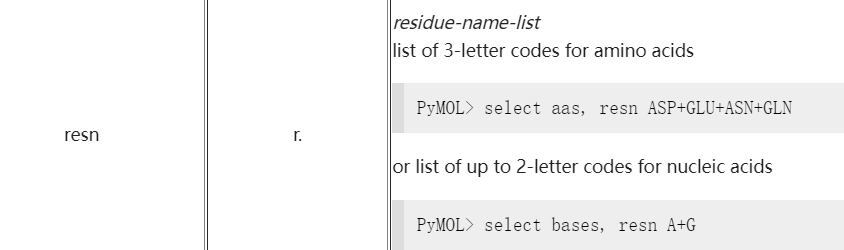
select resn ALA
select resi 100-200
select name CA
select alt A
select index 123
select id 123
select rank 123
select pepseq ACDEF
select label "Hello World"
这里加点我个人的私货:其中select pepseq可以用于抗原表位解析,其实就是高亮蛋白质的特定区域,
chimerax中其实可以直接select——》sequence
标识符匹配(Identifier matching)

select (chain A and resi 100) in (chain A and resi 100 and name CA) # 从链 A 的残基 100 中,选择与目标选择器中完全一致的 α-碳(CA)原子
select (chain A and resi 100) like (chain B and resi 100) # 选择链 A 和链 B 中编号为 100 的残基中名字一致的原子
这里加点我个人的私货:其实相当于是MSA,可以用于大规模蛋白质区域比对之类
实体扩展(Entity expansion)
Entity expansion(实体扩展)是 PyMOL 中的一组操作,用于扩展选择的原子或结构到更高层次的实体,如整个对象、段、链、残基等。它可以帮助用户在选择特定范围的原子后,自动扩展选择范围,以方便进一步操作(如显示、着色等)。主要功能说明:每个操作符的作用是基于原子集合 S1(选择器指定的范围),扩展到更大范围的结构单元。例如,从单个原子扩展到其所属的整个残基或链。需要注意的是: PyMOL 中,所有 “by”-操作符具有弱优先级。例如 byres S1 or S2 会被解析为 byres (S1 or S2),而不是 (byres S1) or S2。
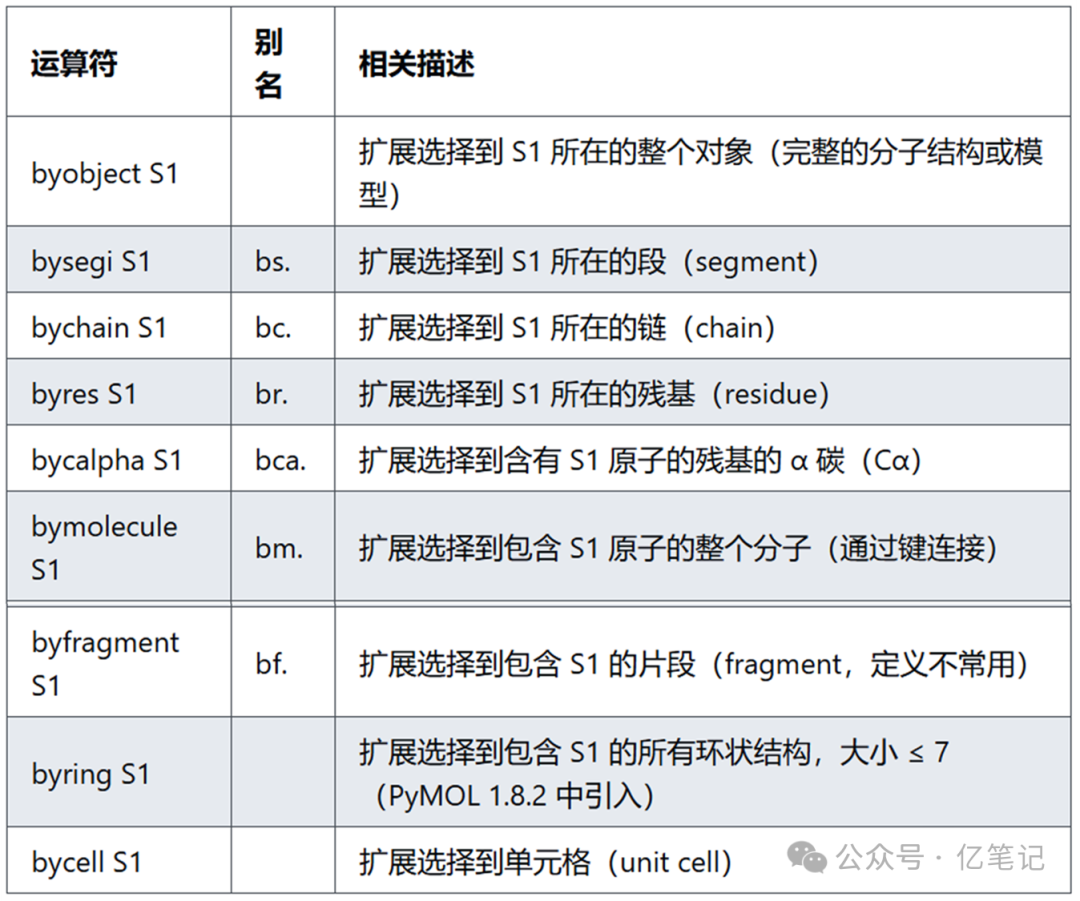
select byobject chain A # 扩展选择到链 A 所在的整个对象(例如,包含链 A 的整个分子结构)
select bs. chain A # 扩展选择到链 A 所在的段(segment)
select bc. resn ALA # 扩展选择到包含残基 ALA 的整个链
select br. name CA # 扩展选择到包含 α 碳(CA)的完整残基
select bca. chain B # 扩展选择到链 B 中所有含有原子的 α 碳(Cα)
select bm. resn ATP # 扩展选择到包含 ATP 残基的整个分子(通过化学键连接的完整分子)
select bf. chain C # 扩展选择到链 C 的所有片段(具体定义取决于分子中的断裂点或碎片划分)
select byring name NZ # 扩展选择到包含原子 NZ 的所有环状结构(限制为 ≤7 原子组成的环)
select bycell chain A # 扩展选择到链 A 所在的单元格(unit cell)
注意注意!!
我个人常用的选择DNA、蛋白质、RNA等结构选择code是
选择蛋白:select protein, (byres polymer & name CA)
选择核酸: select nucleic, (byres polymer & name P)
选择RNA: select rna, (byres polymer & name O2’)
选择DNA: select dna, (nucleic & !rna)
解读如下,因为byres优先级最低,所以byres polymer & name CA实际上是byres (polymer & name CA)其实就是选取聚合物(蛋白质或核酸,pymol中一般不会出现糖)中包含α-C的完整残基
然后核酸中是没有α-C的,所以选择的是蛋白质
细节操作可以参考我之前的博客:
https://blog.csdn.net/weixin_62528784/article/details/143325706
键扩展(Bond expansion)
Bond expansion 操作符是 PyMOL 中用于通过键扩展选择范围的功能。它允许你根据原子之间的化学键关系扩展选定的原子集合。

select bto. resn GLY # 选择所有与 GLY 残基中原子键合的原子,包括 GLY 自己的原子
select nbr. chain A # 选择与链 A 中的原子直接键合的所有原子,但不包括链 A 中的原子本身
select xt. resn ALA # 选择 ALA 残基及其与之直接相连的原子,再进一步扩展 3 个化学键连接的原子
接近度(Proximity)
在 PyMOL 中,Proximity(接近度)操作符用于基于原子之间的距离选择原子。这些操作符可以帮助用户选择位于某一特定距离范围内的原子或分子,例如从某个残基或分子中的原子出发,选择距离它们一定范围内的其他原子。
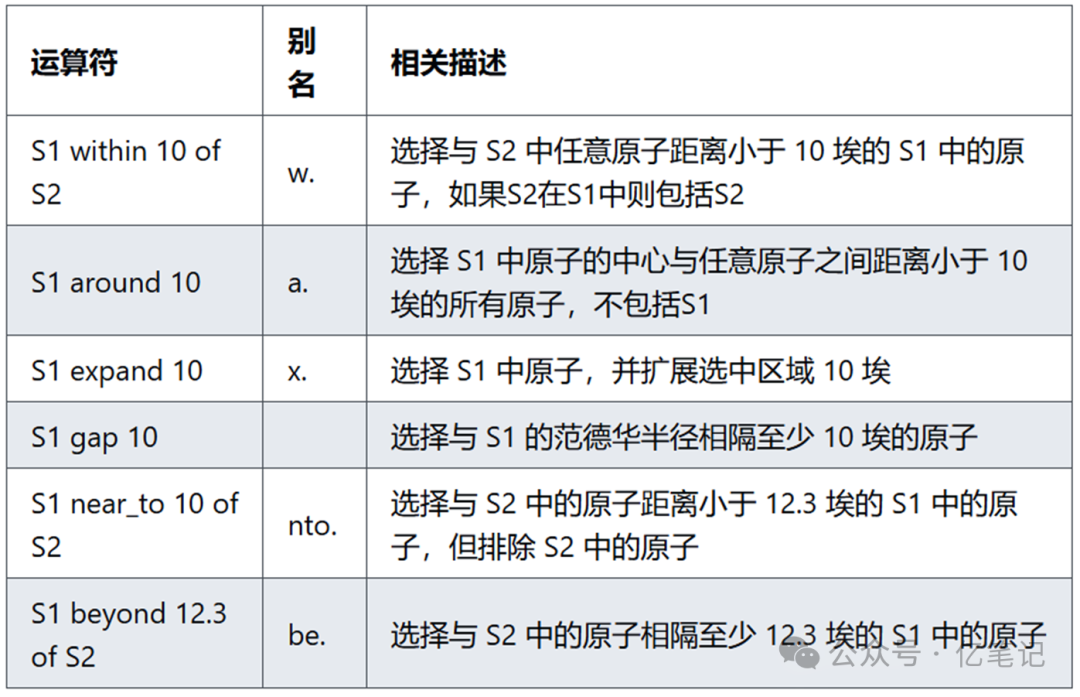
这里倒数第二个勘误是10
select all within 10 of resi 71 # 选择所有与 71 号残基中原子距离小于 10 埃的原子,如果 71 号残基在 all 中则包括 71 号残基
select resi 71 around 10 # 选择以 71 号残基为中心周围 10 埃的原子,一定不包括 71 号残基,与 near_to 等价
select resi 71 expand 10 # 选择以 71 号残基为中心向外扩展 10 埃的原子,一定包括 71 号残基
select resi 71 gap 10 # 选择所有与 71 号残基有至少 10 埃范德华半径间隔的原子
select all near_to 10 of resi 71 # 选择以 71 号残基为中心周围 10 埃的原子,一定不包括 71 号残基,与 around 等价
select all beyond 10 of resi 71 # 选择所有与 71 号残基中原子相隔至少 10 埃的原子
上述几个关于距离选择语法的命令非常接近,下面对这几个命令进行比较。
主要有以下两种语法:
语法一: s1 operator X of s2
语法二: s1 and (s2 operator X)
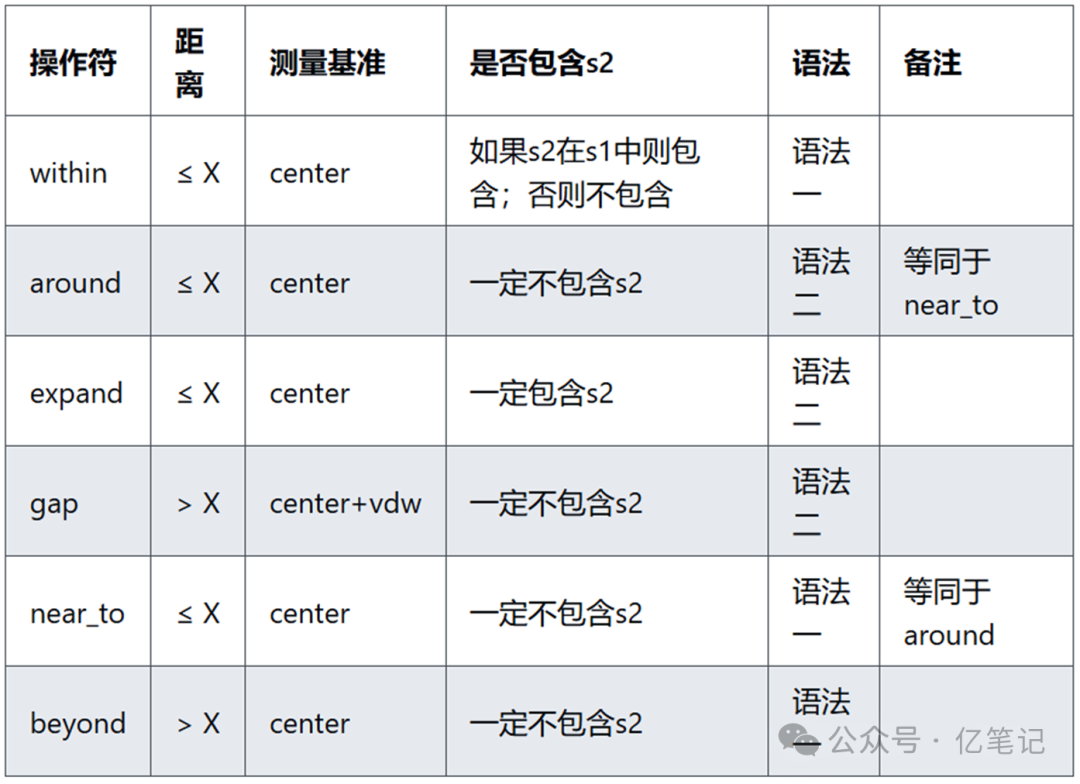
原子属性(Properties)
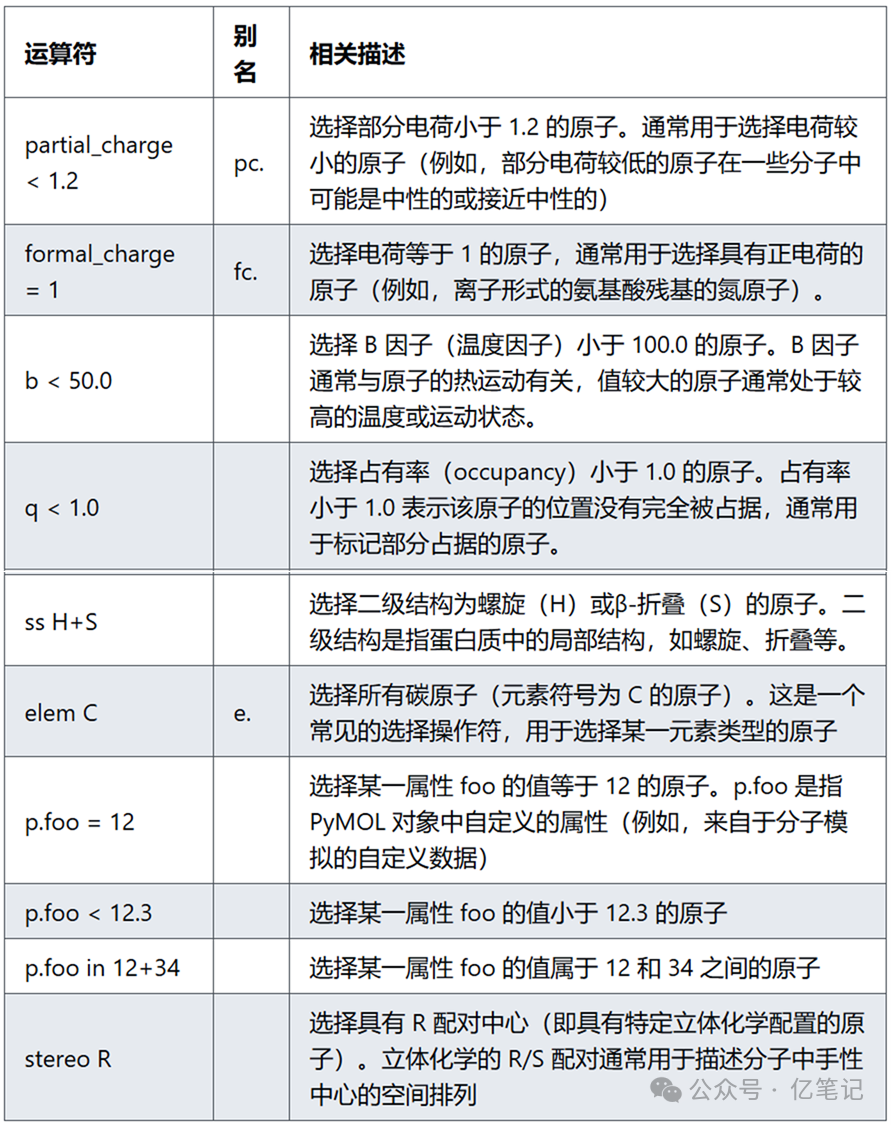
select partial_charge < 1.2 # 选择部分电荷小于 1.2 的原子
select formal_charge < 1 # 选择形式电荷小于 1 的原子
select b < 50 # 选择 B 因子小于 50 的原子
select q < 1 # 选择占有率小于 1 的原子
select ss H+S # 选择在螺旋或 β-折叠结构中的原子
select elem C # 选择所有碳原子
select p.foo = 12 # 选择所有 foo 属性值为 12 的原子
select p.foo < 12.3 # 选择所有 foo 属性值小于 12.3 的原子
select p.foo < 12+34 # 选择所有 foo 属性值在 12 到 34 之间的原子
select stereo R # 选择所有 R 配对的手性原子
标志(Flags)
用于选择具有特定“标志”或状态的原子的操作符。标志(flags)通常是指原子的某些特定状态或附加属性。
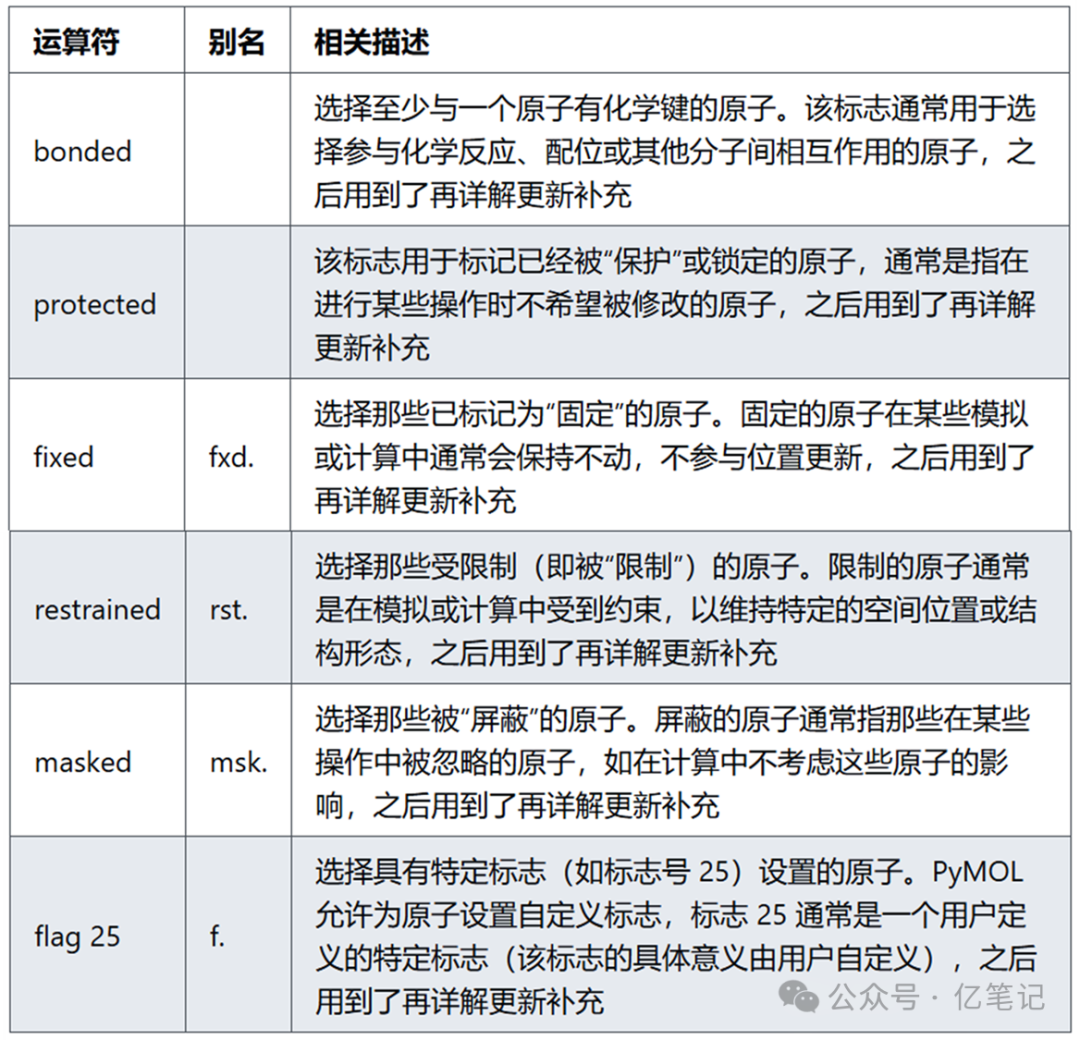
select bonded # 选择所有与至少一个原子有键的原子
select protected # 选择所有被“保护”的原子。通常需要通过其他命令(如 protect)来设置保护状态
select fixed # 选择所有固定的原子。通常通过其他命令(如 fix)来设置固定状态
select restrained # 选择所有被限制的原子。通常通过其他命令(如 restrain)来设置限制
select masked # 选择所有被屏蔽的原子。通常通过其他命令(如 mask)来设置屏蔽状态
select flag 25 # 选择所有标志 25 的原子。标志 25 通常是由用户在脚本或命令中设置的,具体含义需要参考脚本中的设定。
化学类(Chemical classes)
选择不同类型的原子或分子。使用这些类,可以选择所有水分子、蛋白质、核酸、配体、金属离子等,甚至选择聚合物的侧链或骨架部分。PyMOL通过这些分类帮助用户对结构进行精细选择,尤其是在处理复杂分子或蛋白质结构时。
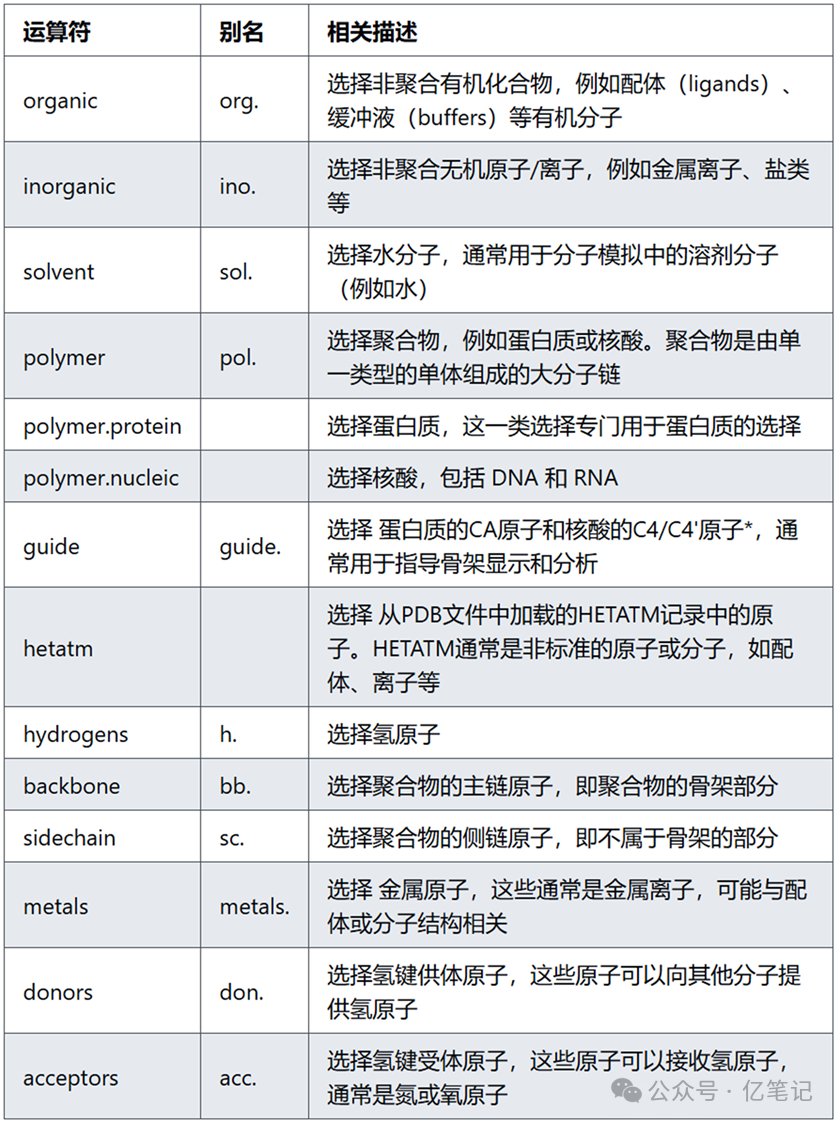
select organic # 选择所有的有机分子,这些分子可能是实验中的药物分子、配体或其他小分子
select inorganic # 选择所有的无机原子或离子,通常包括金属离子(如钠、钙、镁等)或者其他无机元素
select solvent # 选择所有的水分子(H2O),这些分子在分子动力学模拟中通常作为溶剂存在
select polymer # 选择所有的聚合物,包括蛋白质或核酸分子
select polymer.protein # 选择所有的蛋白质分子,蛋白质是由氨基酸组成的聚合物
select polymer.nucleic # 选择所有的核酸分子,主要包括 DNA 和 RNA 分子
select guide # 选择所有蛋白质的α碳(CA)原子和核酸的C4*(DNA)或C4'(RNA)原子,通常用于分析分子的主链结构
select hetatm # 选择所有HETATM记录中的原子,通常用于选择配体、金属离子等非标准分子
select hydrogens # 选择所有的氢原子,这在处理分子时有助于选择氢键或者其他分子间的相互作用
select backbone # 选择所有聚合物的主链原子,通常用于蛋白质或核酸的骨架分析
select sidechain # 选择所有聚合物的侧链原子,通常用于蛋白质的侧链分析
select metals # 选择所有的金属原子,通常是金属离子,如锌、镁、钙等
select donors # 选择所有氢键供体原子,用于分析氢键形成
select acceptors # 选择所有氢键受体原子,用于分析氢键形成
注意注意!!
我个人常用的选择DNA、蛋白质、RNA等结构选择code是
选择蛋白:select protein, (byres polymer & name CA)
选择核酸: select nucleic, (byres polymer & name P)
选择RNA: select rna, (byres polymer & name O2’)
选择DNA: select dna, (nucleic & !rna)
解读如下,因为byres优先级最低,所以byres polymer & name CA实际上是byres (polymer & name CA)其实就是选取聚合物(蛋白质或核酸,pymol中一般不会出现糖)中包含α-C的完整残基
然后核酸中是没有α-C的,所以选择的是蛋白质。
然后对照上面的这一段指示,其实就更加容易理解
当然,除了使用polymer,可以直接使用蛋白质的polymer.protein或者是polymer.nucleic
这样修改之后的
#选择蛋白质:select protein, byres polymer.protein
#选择核酸:select nucletic, byres polymer.nucleic
#选择DNA:如果仅仅只是按照上面的方法来,也是简单的
#选择RNA:如果仅仅只是按照上面的 name O2’,那么嵌套是简单的
可以看到,确实效果上对于蛋白质是符合的、一致的

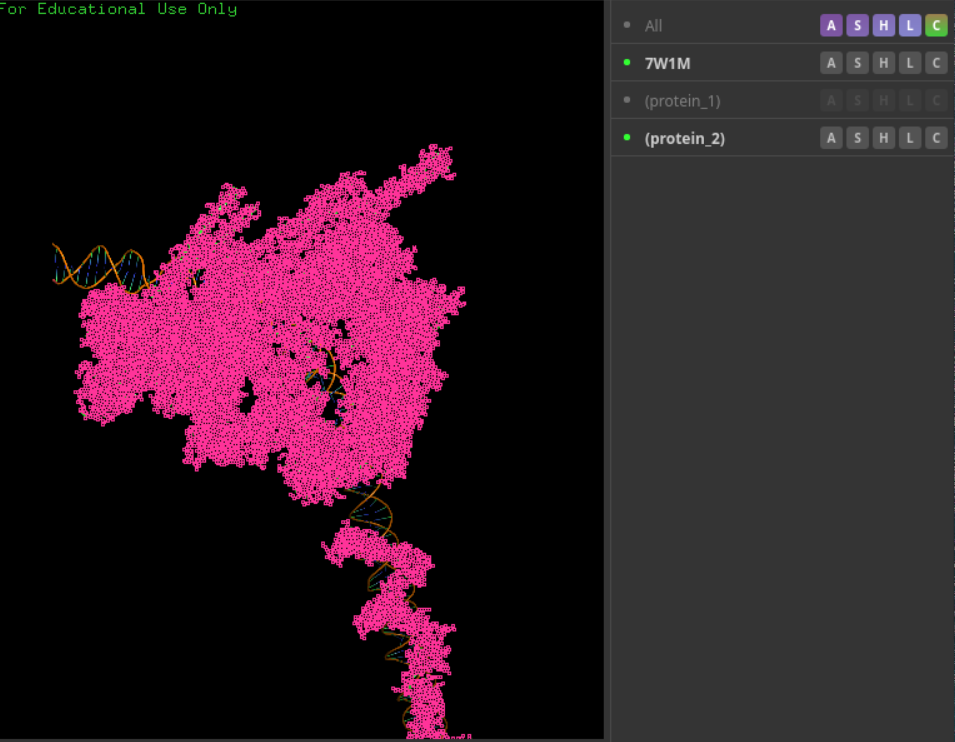
然后对于核酸:确实也是一致的、符合的
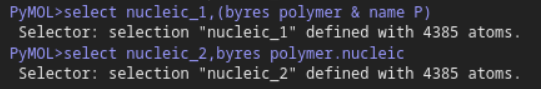
但是直接对DNA或者是RNA选择:
比如说DNA,从原来方法看,貌似都是间接的:
需要从nucleic中去除RNA,
而RNA可以直接从nucleic中选择,或者是从原始polymer中选择;
而nucleic,可以直接使用polymer.nucleic,或者是从polymer中获取。
有没有方法可以直接获取DNA或者是RNA?
貌似没有最有效的方法?
那你们知道我所提供的最原始的生物大分子选择方法是哪里来的吗?
https://sourceforge.net/p/pymol/mailman/message/36149679/

我之前也想过要使用直接从resn上获取核酸的8个字符
resn A+C+G+U+DA+DC+DG+DT
但实际上这种方法是不行的,可能会遗漏一些最基本的核酸修饰

另外可以参考:https://pymolwiki.org/index.php/Property_Selectors
综上,除了优化nucleic,其余的都参考最原始的方案
样式(Style)
用于根据可视化的表示样式或属性选择原子。通过这些选择符,用户可以根据不同的样式属性来选择分子中与这些样式相关的原子。这些 样式选择符(Style) 使得用户可以基于可视化样式或属性(如是否可见、卡通表示、颜色设置等)来选择特定的原子。
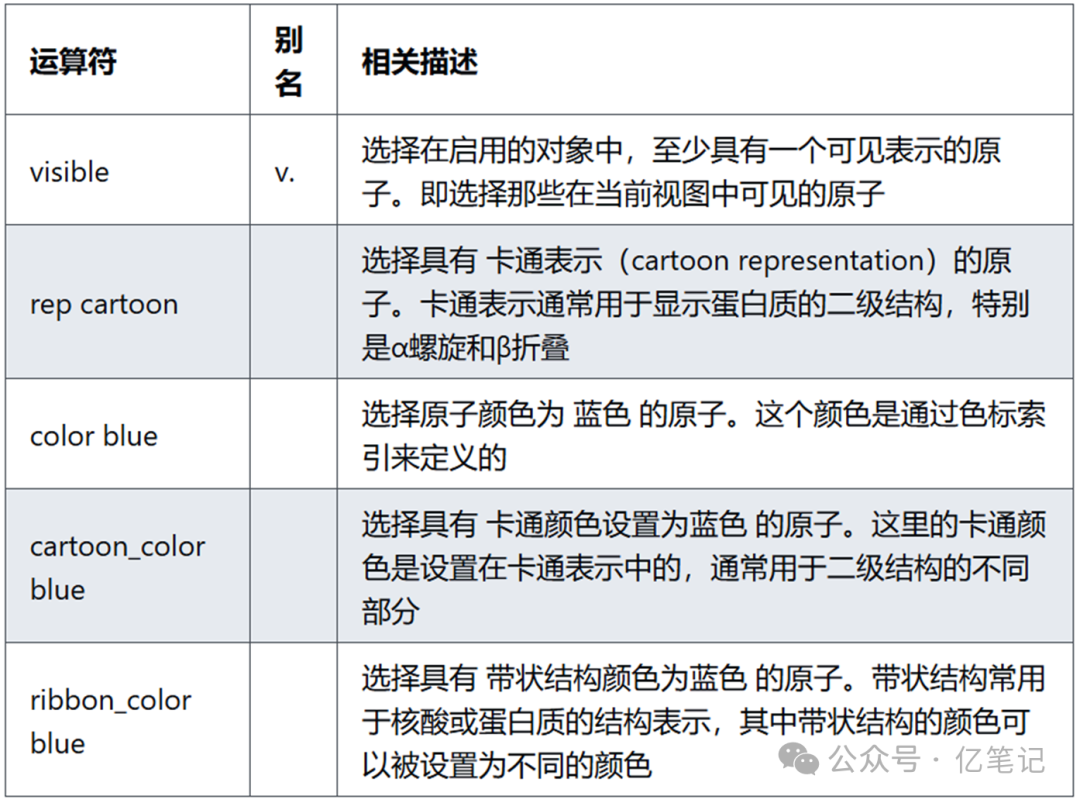
select visible # 选择所有具有可见表示的原子,通常用于选择当前可视化中显示的原子
select rep cartoon # 选择所有具有卡通表示的原子,常用于蛋白质二级结构的可视化
select color blue # 选择所有颜色为蓝色的原子,通常用于选择那些在当前可视化中设置为蓝色的原子
select cartoon_color blue # 选择所有卡通表示中颜色为蓝色的原子,常用于选择卡通表示中着色为蓝色的部分
select ribbon_color blue # 选择所有带状结构中颜色为蓝色的原子,常用于显示在带状结构中设置为蓝色的部分
伪原子(Non molecular)
用于选择“非分子”类型的伪原子,这些伪原子不是实际的分子原子,而是用来帮助控制视图、定位或旋转等。center 代表场景的几何中心,通常用于调整视图,确保目标分子或结构位于可视化窗口的中心。origin 代表旋转的原点,它是视图旋转的参考点,用于控制视角和旋转。

zoom center
zoom origin
坐标(Coordinates)
用来选择与原子坐标相关的原子。这些选择符可以通过原子的空间坐标(如 x, y, z)来筛选原子。
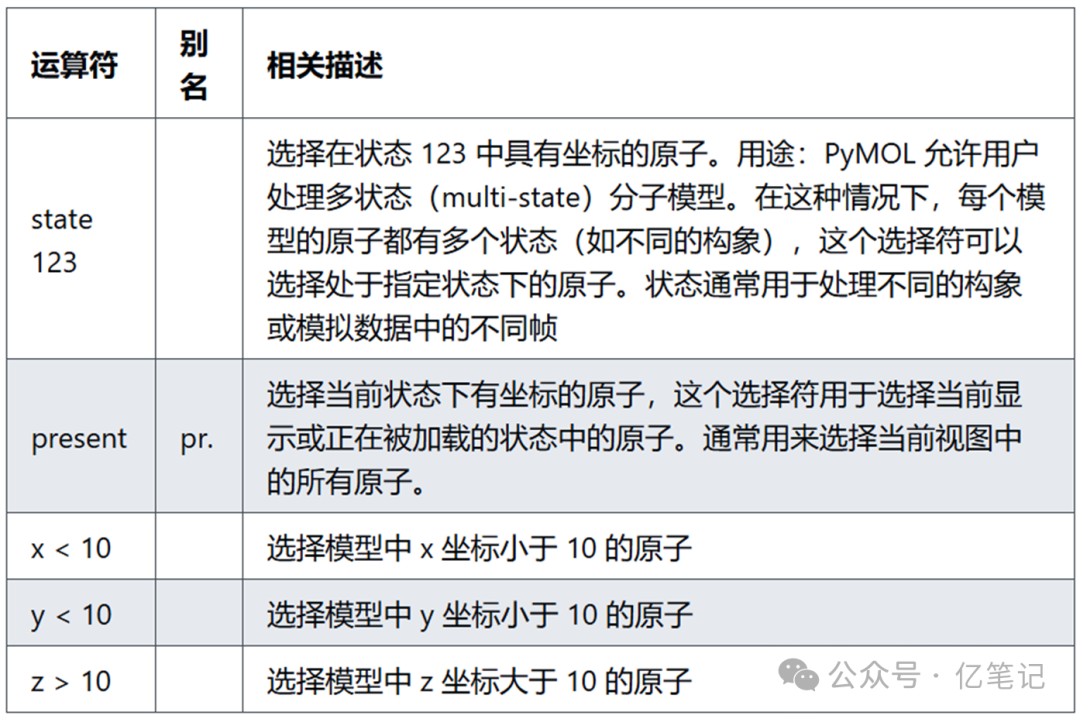
select state 123 # 选择状态为 123 的所有原子
select present # 选择当前状态下的所有原子
select x < 10 # 选择所有 x 坐标小于 10 埃的原子
select y < 10 # 选择所有 y 坐标小于 10 埃的原子
select z > 10 # 选择所有 z 坐标大于 10 埃的原子
原子类型(Atom typing)
根据原子的类型进行选择。
select text_type TT # 选择具有 text_type 类型的所有原子,适用于与文本标签相关的原子
select numeric_type 123 # 选择所有具有数字类型标识的原子,适用于某些数值属性的自动分配或标识
参考博客中常用的一些选择语句:
https://mp.weixin.qq.com/s/zIxz0caZgFnTulQOMVekHg
md.select("chain A") # 选择蛋白的某一条链
cmd.select("resn F90") # F90是残基名称,选择某个残基
cmd.select("(br. all within 1 of resn %s) and (not resn %s)"%(i[2],i[2])) # 选择某残基一定距离范围内的所有残基
cmd.select("(model %s and resn %s) or (model %s)"%(i[1], i[2], i[0])) # 选择一个object中的一个残基和另一个object
cmd.select("(br. all within 4 of (mode object_a or mode object_b))") # 3个object (object_a, object_b, object_c),寻找 object_c 上与 object_a 和 object_b 残基距离在4埃内的残基
cmd.select("resi 1-7 and mode object_b") # 选择某个object中的一段连续残基
cmd.select("resi 3-7+23-34+45-49 and lig_1.pdb") # 选择某个object中的多段残基
cmd.select("resi 1 and (name OP1 or name P or name OP2)") # 选择残基上某些特定原子
cmd.select("polymer.nucleic within 4 of polymer.protein") # 选择蛋白4埃范围内的所有RNA原子,选择一个object附近一定距离内的所有其他object的所有原子
参考:
https://pymolwiki.org/index.php/Selection_Algebra
https://www.compchems.com/pymol-selection-tool/#combining-selection-rules-with-logical-operators
https://pymol.org/dokuwiki/doku.php?id=selection
三,chimeraX的命令行处理例子:
1,如何使用chimeraX的样式命令来改变分子的显示模式,包括球棍、线框、表面和卡通等?
如何使用剪切面来切割分子,以及如何使用透明度来调节分子的可视化效果?
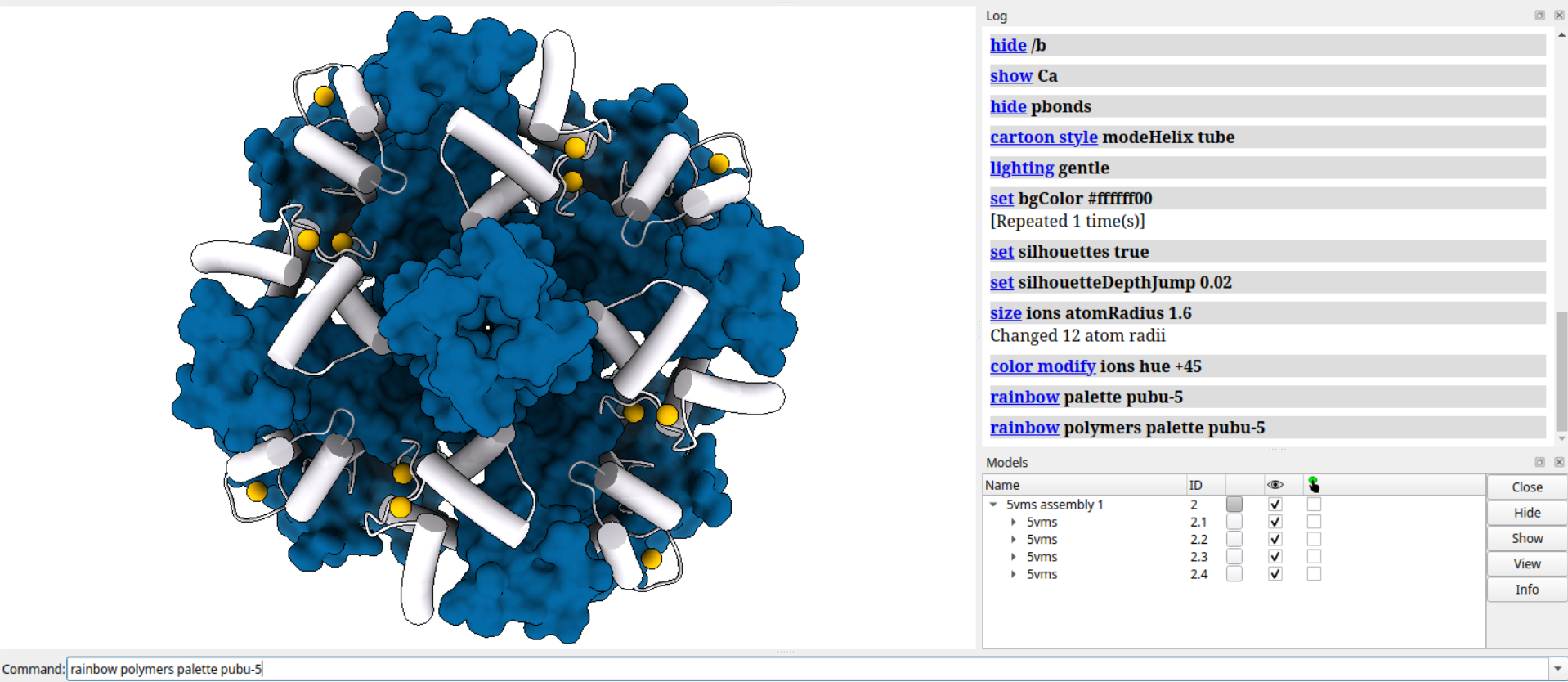
open 5vms
sym #1 assembly 1 copies true
close #1
view orient
zoom 1.1
turn x 180
surf /a
hide /a cartoons
hide /b
show Ca
hide pbonds
cartoon style modeHelix tube
lighting gentle
set bgColor #ffffff00
set silhouettes true
set silhouettedepth 0.02
size ions atom 1.6
color modify ions hue +45
rainbow palette pubu-5
rainbow polymers palette pubu-5
#open 5vms: 打开名为5vms的模型文件
#sym: 1 assembly 1 copies true: 显示模型#1的对称性拷贝,根据PDB文件中的assembly 1信息
#close: 1: 关闭模型#1,只保留它的对称性拷贝
#view orient: 将视角调整为正交投影,使得模型在屏幕上居中显示
#zoom 1.1: 将视角放大10%,使得模型看起来更大
#turn x 180: 将模型绕着x轴旋转180度,使得它上下颠倒
#surf /a: 为所有的a链生成表面模型
#hide /a cartoons: 隐藏所有的a链的卡通模型,只显示表面模型
#hide /b: 隐藏所有的b链,不显示任何模型
#show Ca: 显示所有的Ca原子,即所有氨基酸残基的α碳原子
#hide pbonds: 隐藏所有的假键,即连接不同残基或分子的键
#cartoon style modeHelix tube: 将卡通模型中的α螺旋显示为管状,而不是带状
#lighting gentle: 将光照效果设置为柔和模式,使得表面看起来更平滑
#set bgColor: ffffff00: 将背景颜色设置为白色,其中#ffffff00是十六进制的颜色代码
#set silhouettes true: 开启轮廓效果,使得模型边缘更清晰
#set silhouettedepth 0.02: 将轮廓效果的深度设置为0.02,即轮廓线的宽度与屏幕高度的比例
#size ions atom 1.6: 将离子原子(如Na+或Cl-)的大小设置为1.6埃(Å),即与氢原子相同
#color modify ions hue +45: 将离子原子的颜色按色相(hue)增加45度,使得它们看起来更鲜艳
#rainbow palette pubu-5: 将彩虹色彩方案设置为pubu-5,即紫蓝色调的五种颜色
#rainbow polymers palette pubu-5: 将蛋白质或核酸链按照彩虹色彩方案着色,即每条链从一端到另一端渐变颜色
#chimeraX
# 打开PDB ID为5vms的蛋白质结构数据
open 5vms
# 对#1模型应用对称操作,生成装配体1的所有拷贝
sym #1 assembly 1 copies true
# 关闭#1模型
close #1
# 重新调整视图,使其居中并适合屏幕
view orient
# 放大视图1.1倍
zoom 1.1
# 绕x轴旋转180度
turn x 180
# 对链A生成表面
surf /a
# 隐藏链A的卡通表示
hide /a cartoons
# 隐藏链B
hide /b
# 显示Cα原子
show Ca
# 隐藏假键
hide pbonds
# 将卡通表示的螺旋样式设置为管状
cartoon style modeHelix tube
# 设置柔和的光照效果
lighting gentle
# 设置背景颜色为透明白色
set bgColor #ffffff00
# 启用轮廓线
set silhouettes true
# 设置轮廓线深度为0.02
set silhouettedepth 0.02
# 设置离子的原子大小为1.6
size ions atom 1.6
# 修改离子的颜色,色调增加45度
color modify ions hue +45
# 使用pubu-5调色板对彩虹颜色进行设置
rainbow palette pubu-5
# 使用pubu-5调色板对聚合物进行彩虹颜色设置
rainbow polymers palette pubu-5
打开的是5VMS
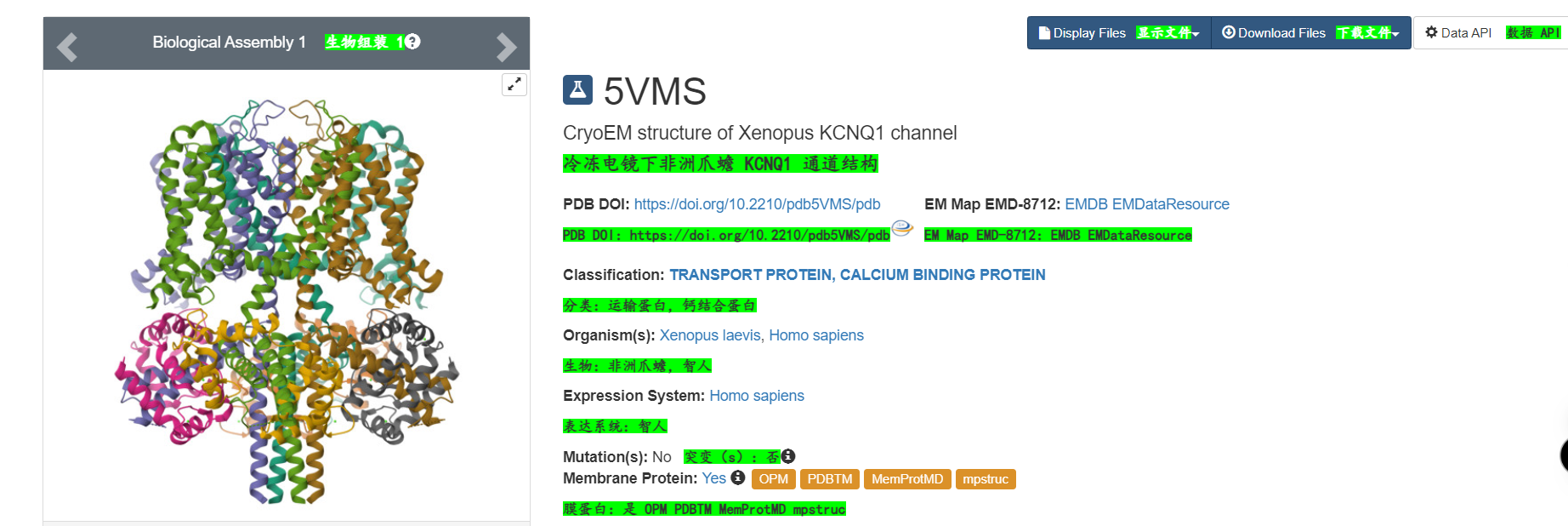
从下面这一步开始操作的每一步的效果,注意命令行都在下面:
# 打开PDB ID为5vms的蛋白质结构数据
open 5vms
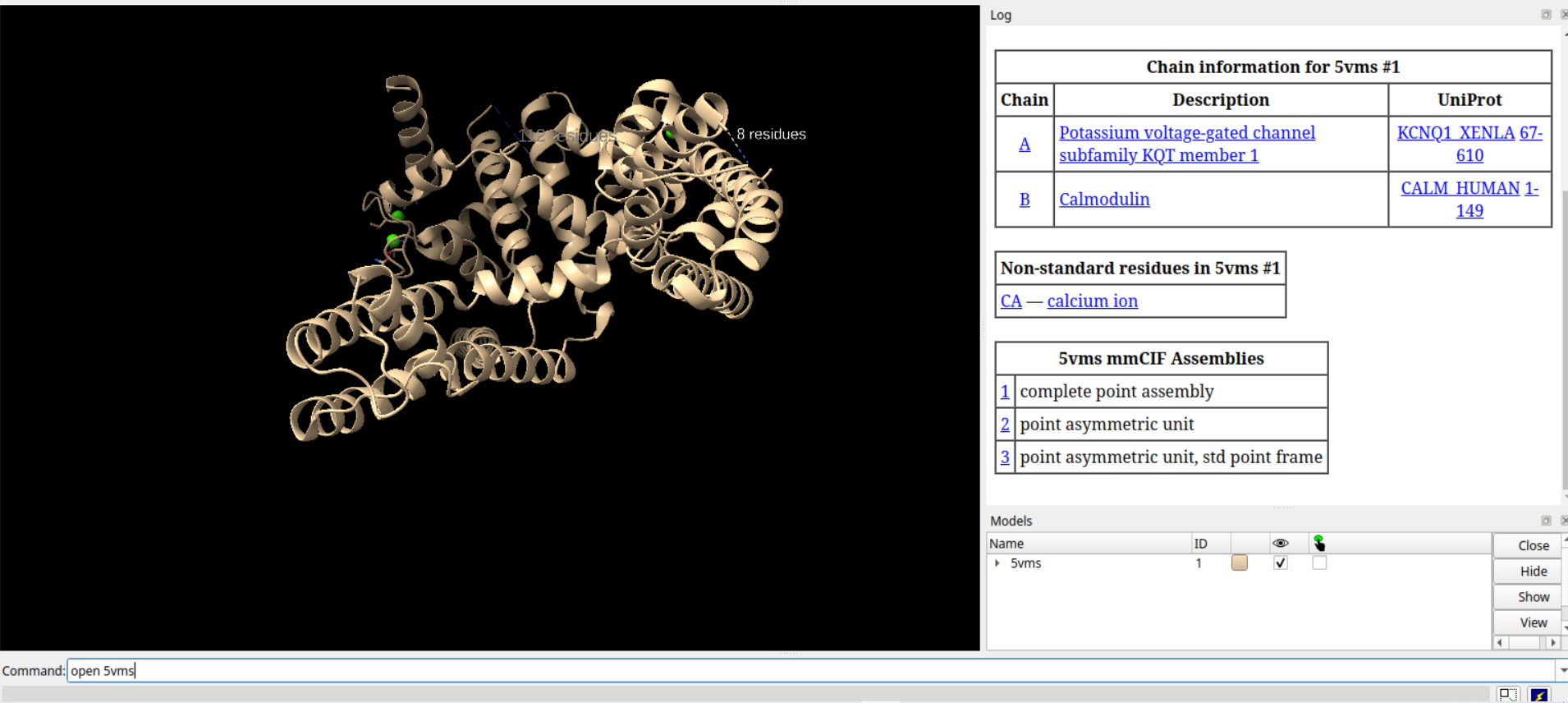
# 对#1模型应用对称操作,生成装配体1的所有拷贝
sym #1 assembly 1 copies true
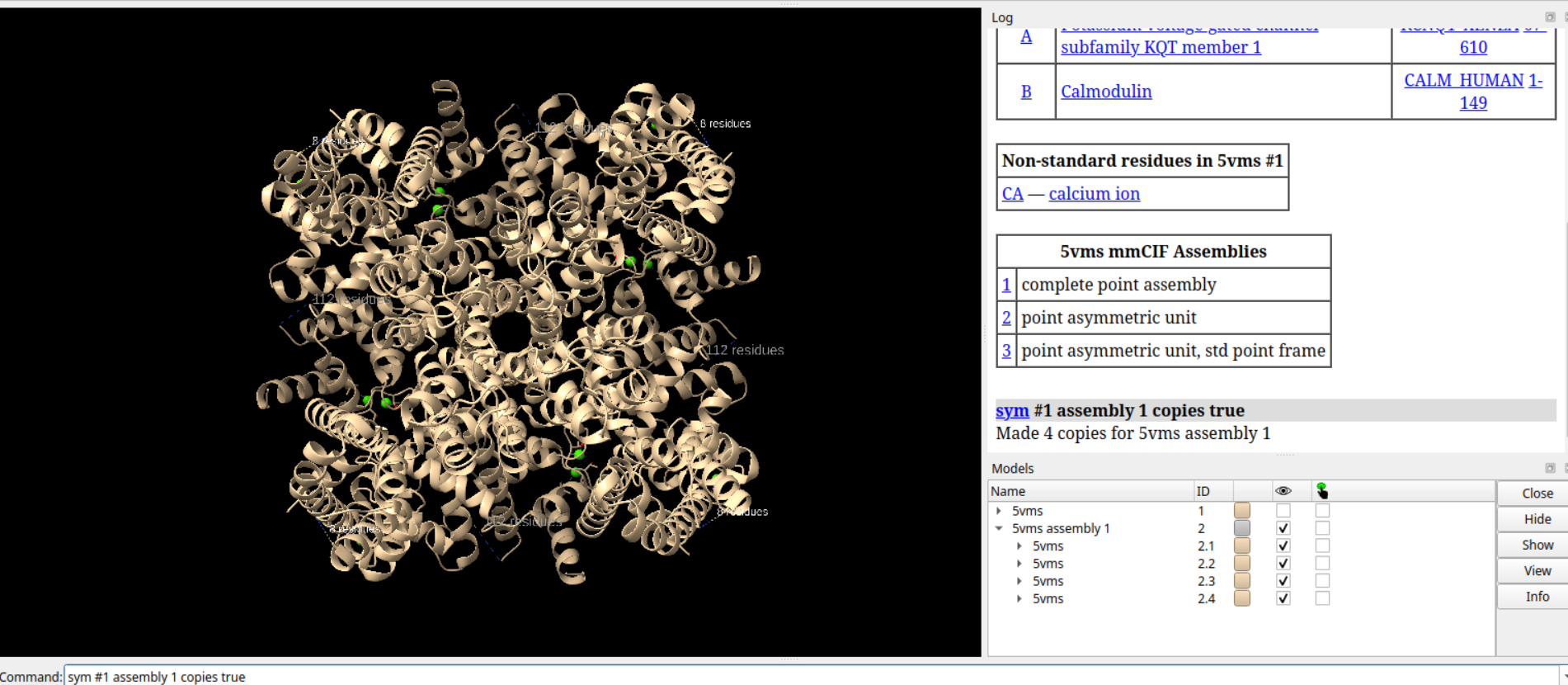
# 关闭#1模型
close #1
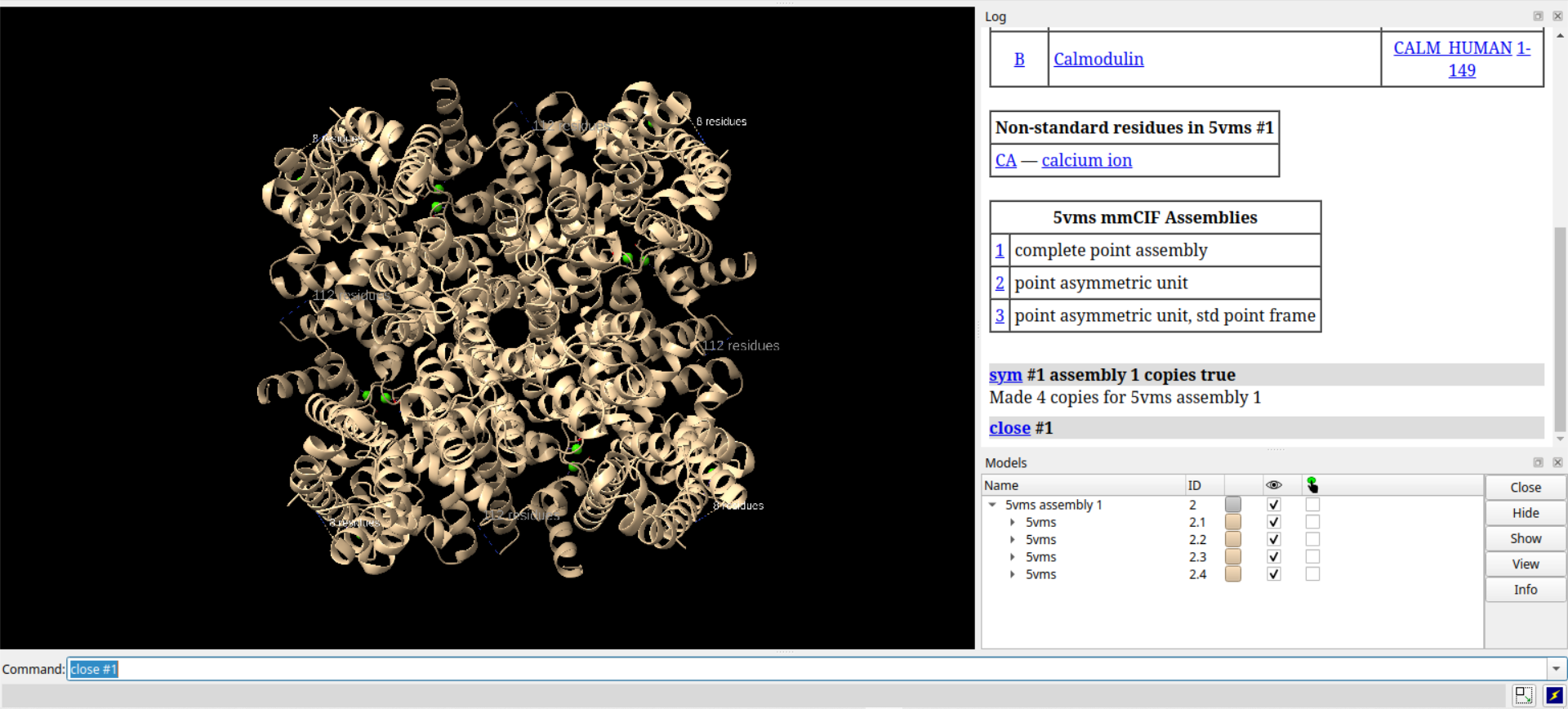
# 重新调整视图,使其居中并适合屏幕
view orient
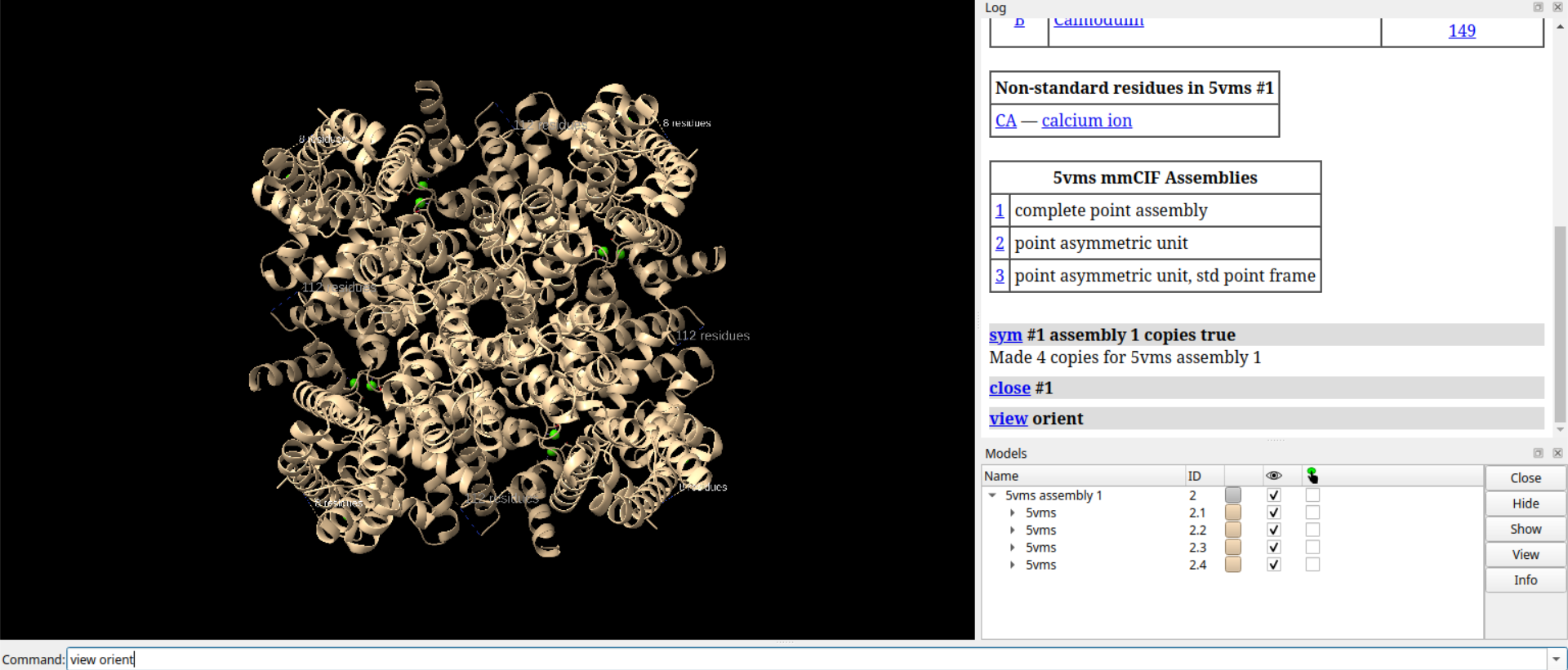
# 放大视图1.1倍
zoom 1.1
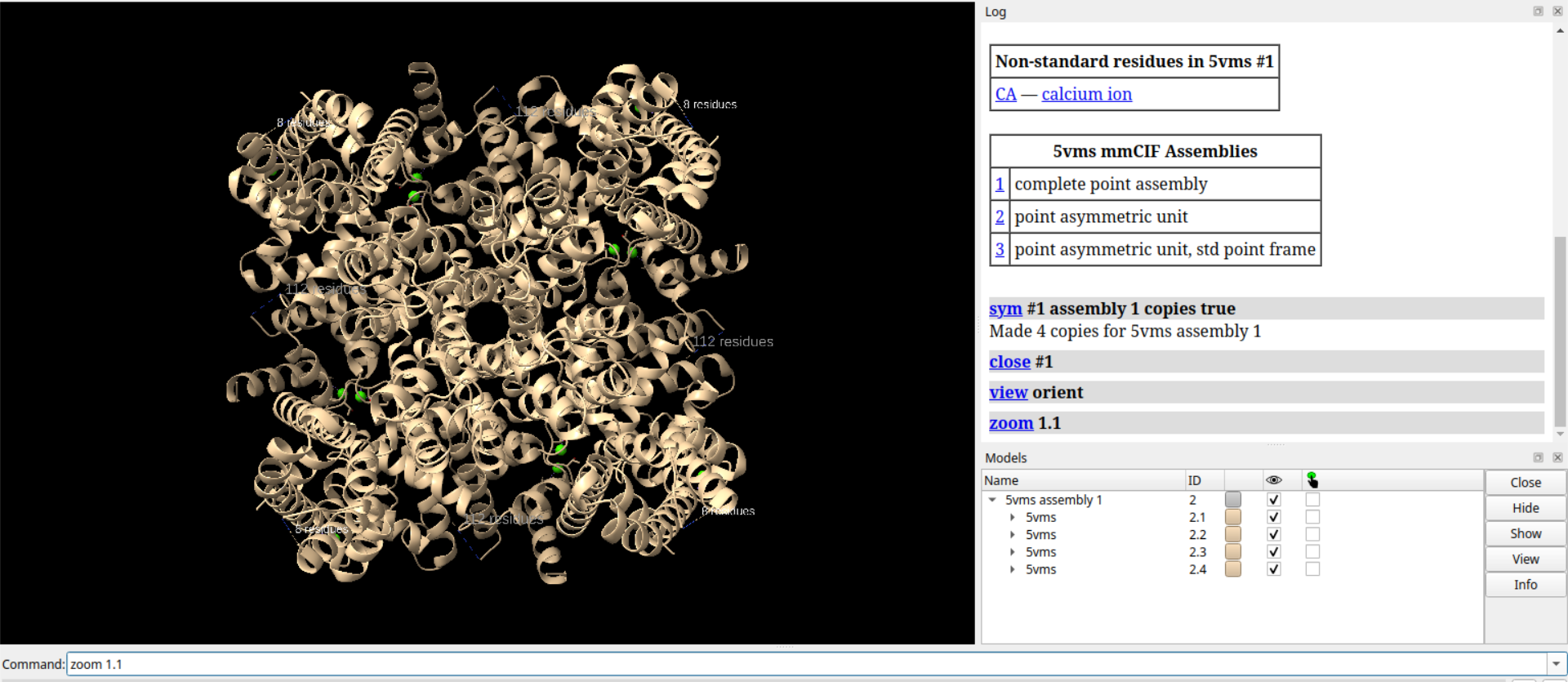
# 绕x轴旋转180度
turn x 180
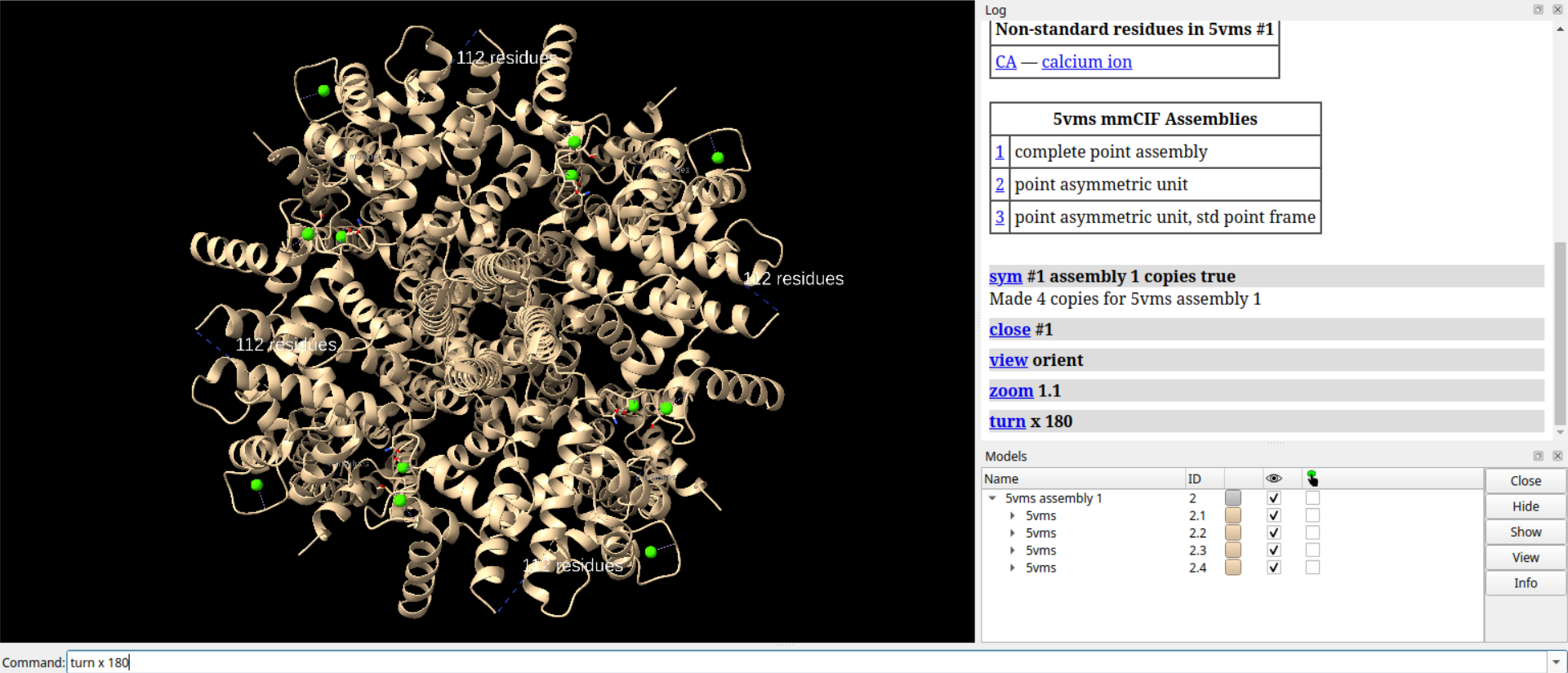
# 对链A生成表面
surf /a
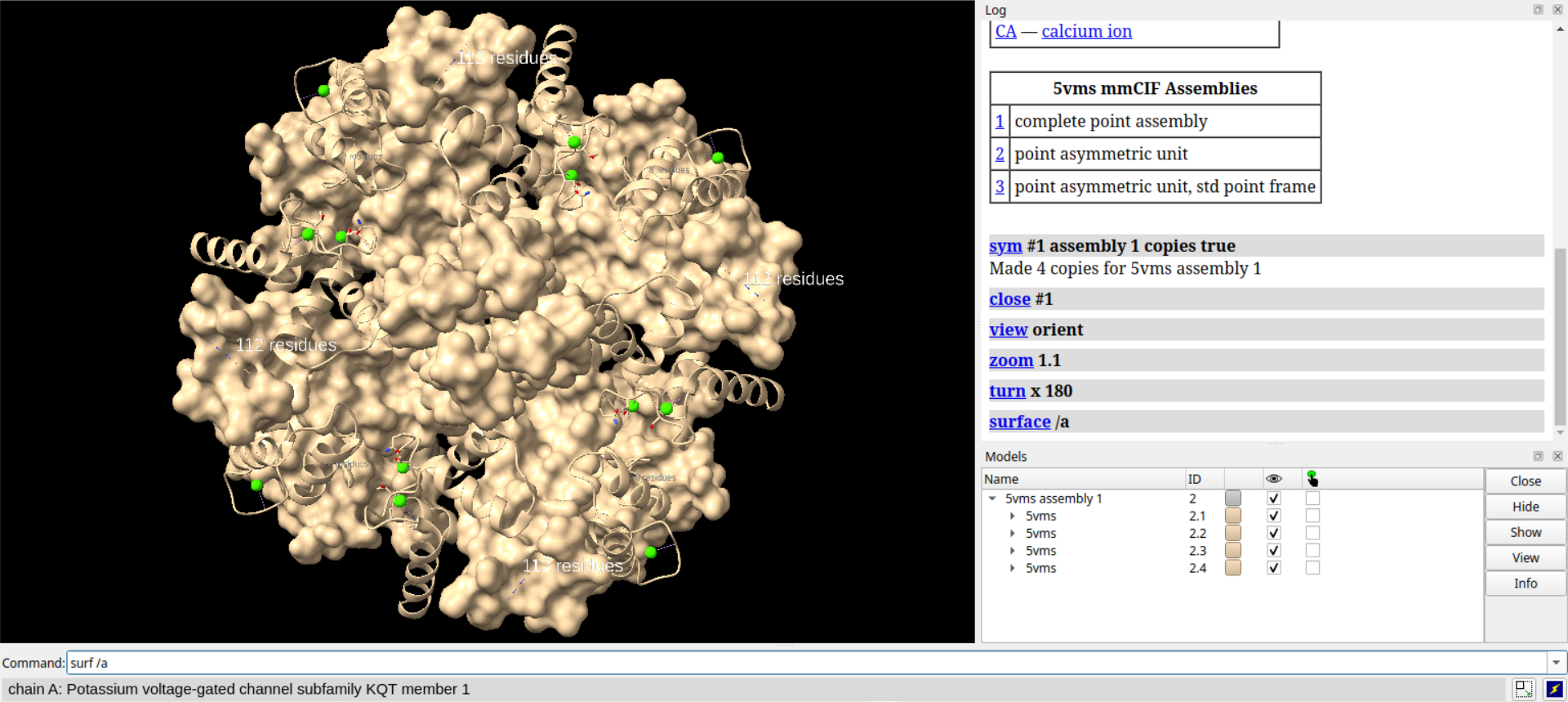
# 隐藏链A的卡通表示
hide /a cartoons
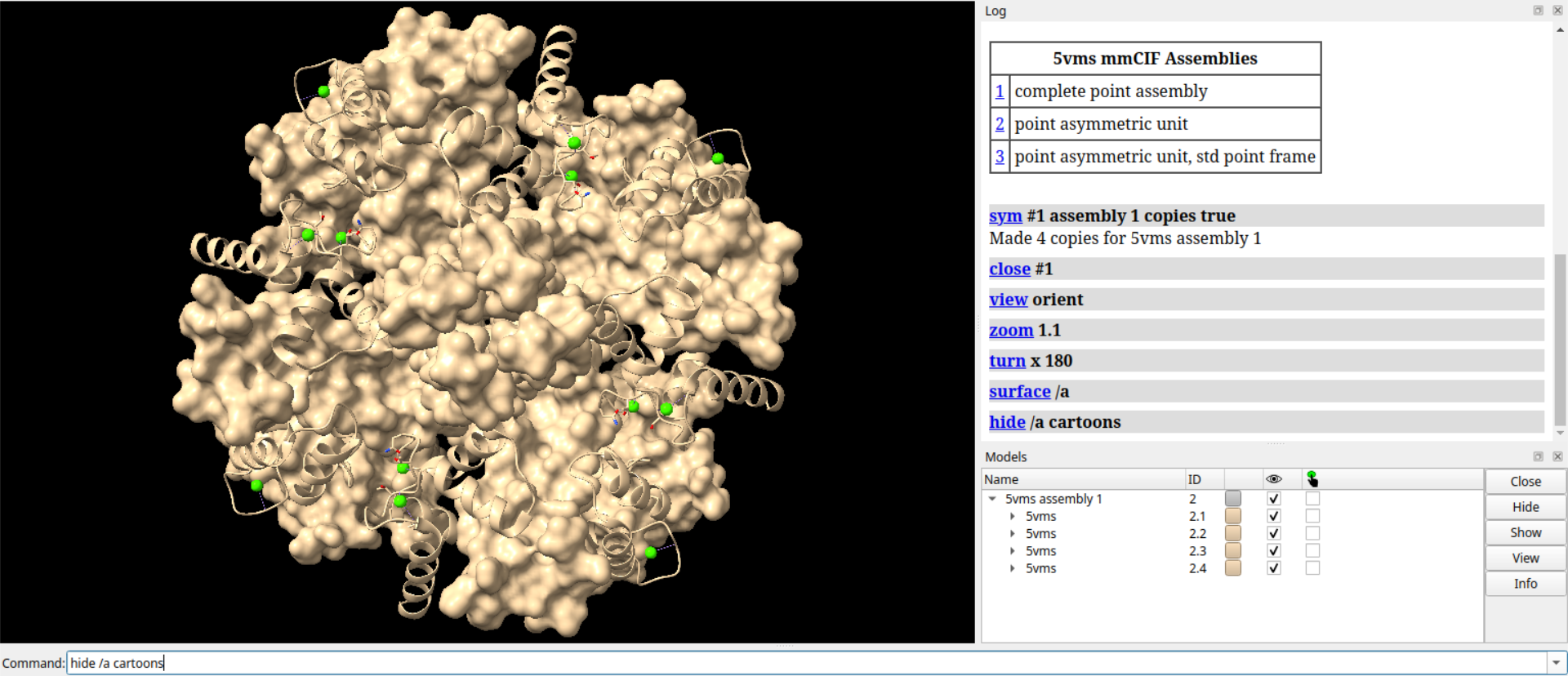
# 隐藏链B
hide /b
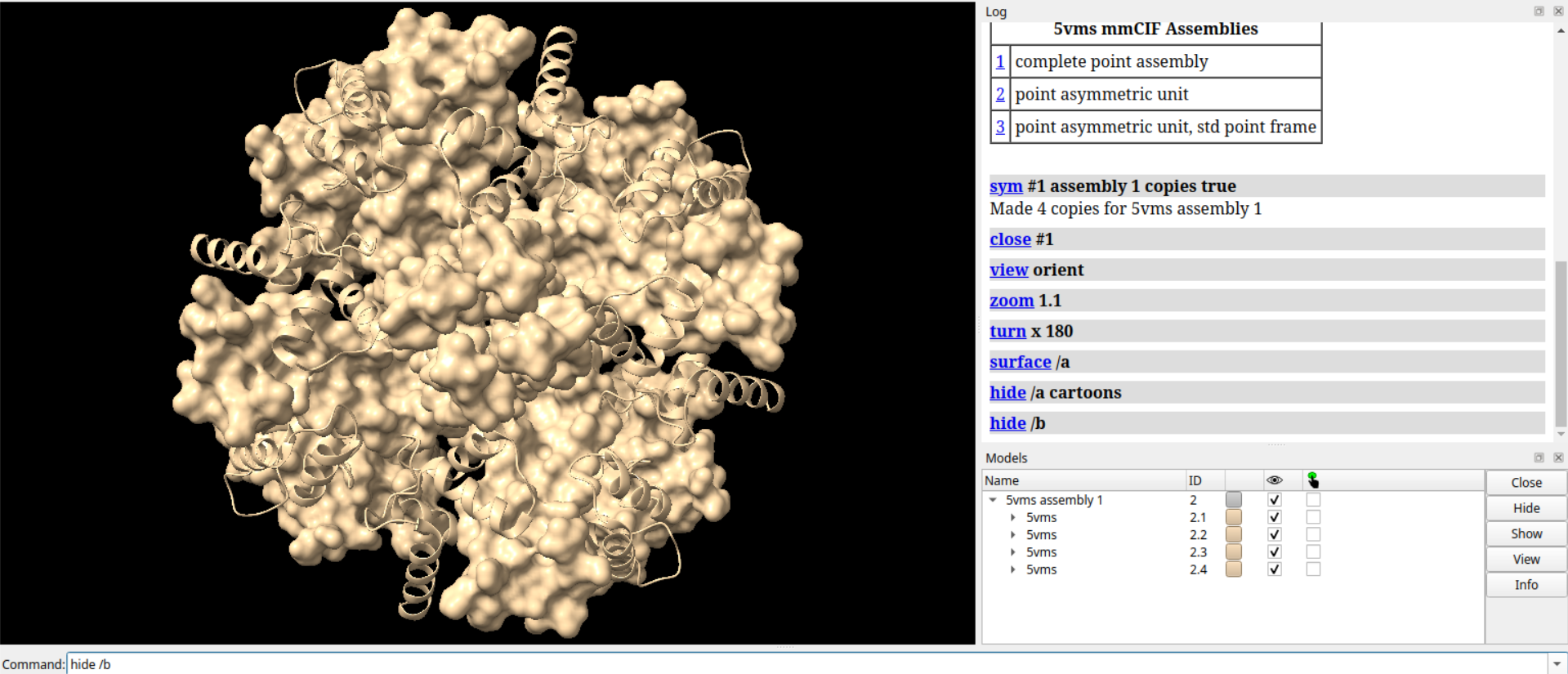
# 显示Cα原子
show Ca
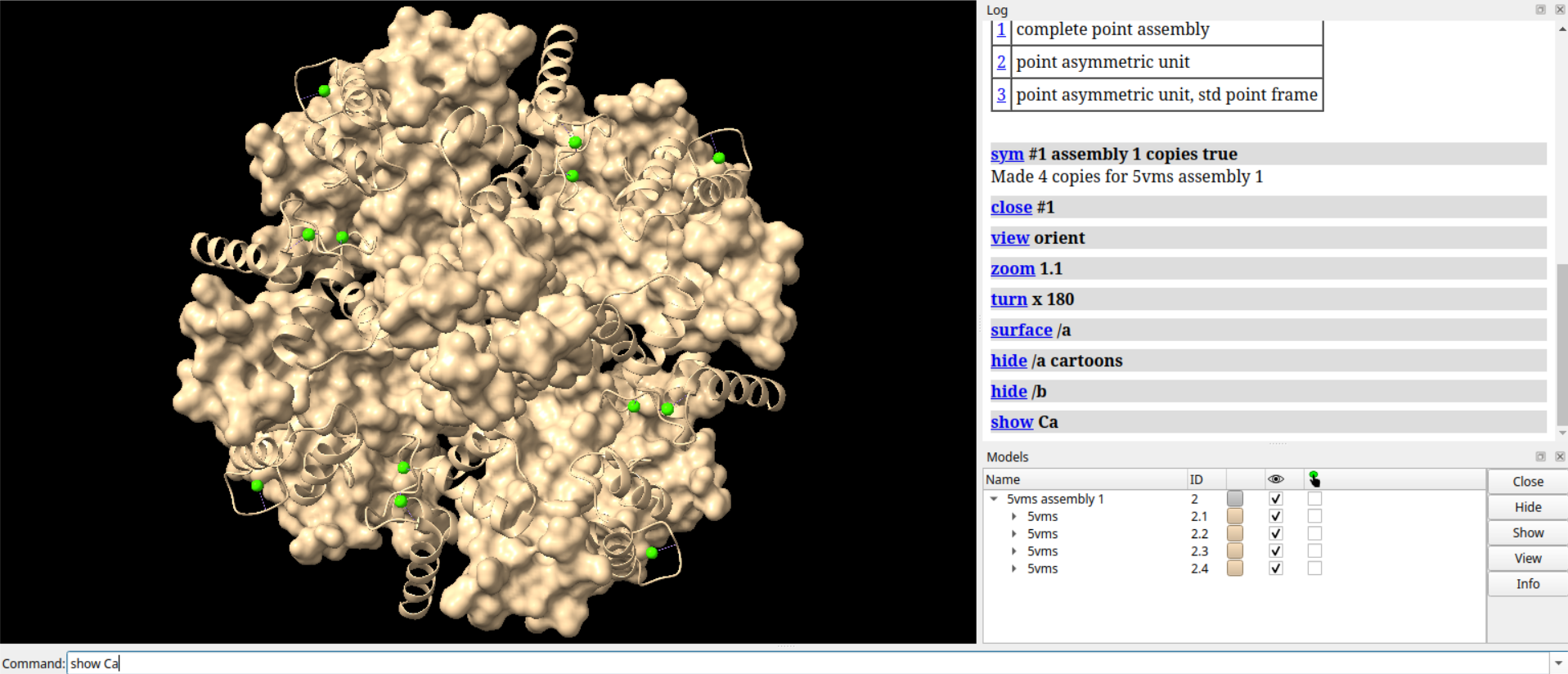
# 隐藏假键
hide pbonds
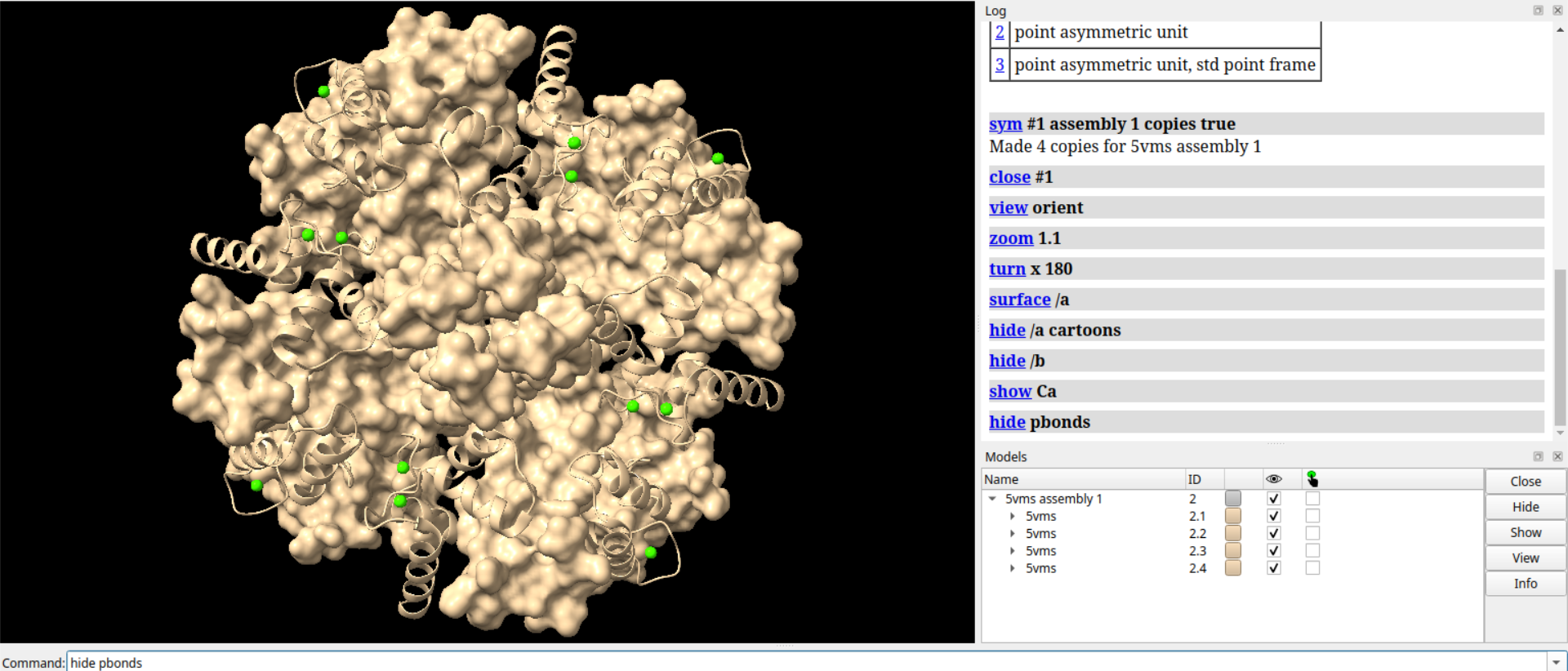
# 将卡通表示的螺旋样式设置为管状
cartoon style modeHelix tube
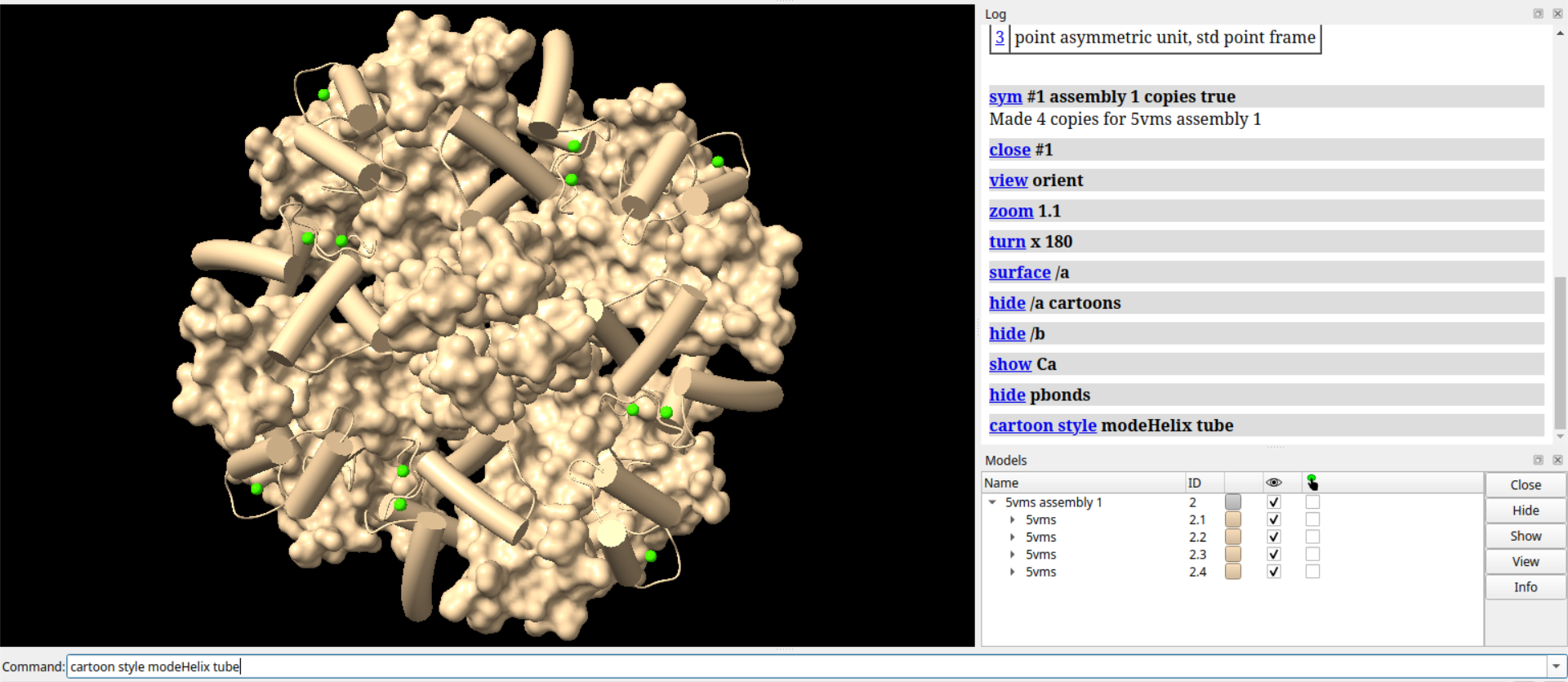
# 设置柔和的光照效果
lighting gentle
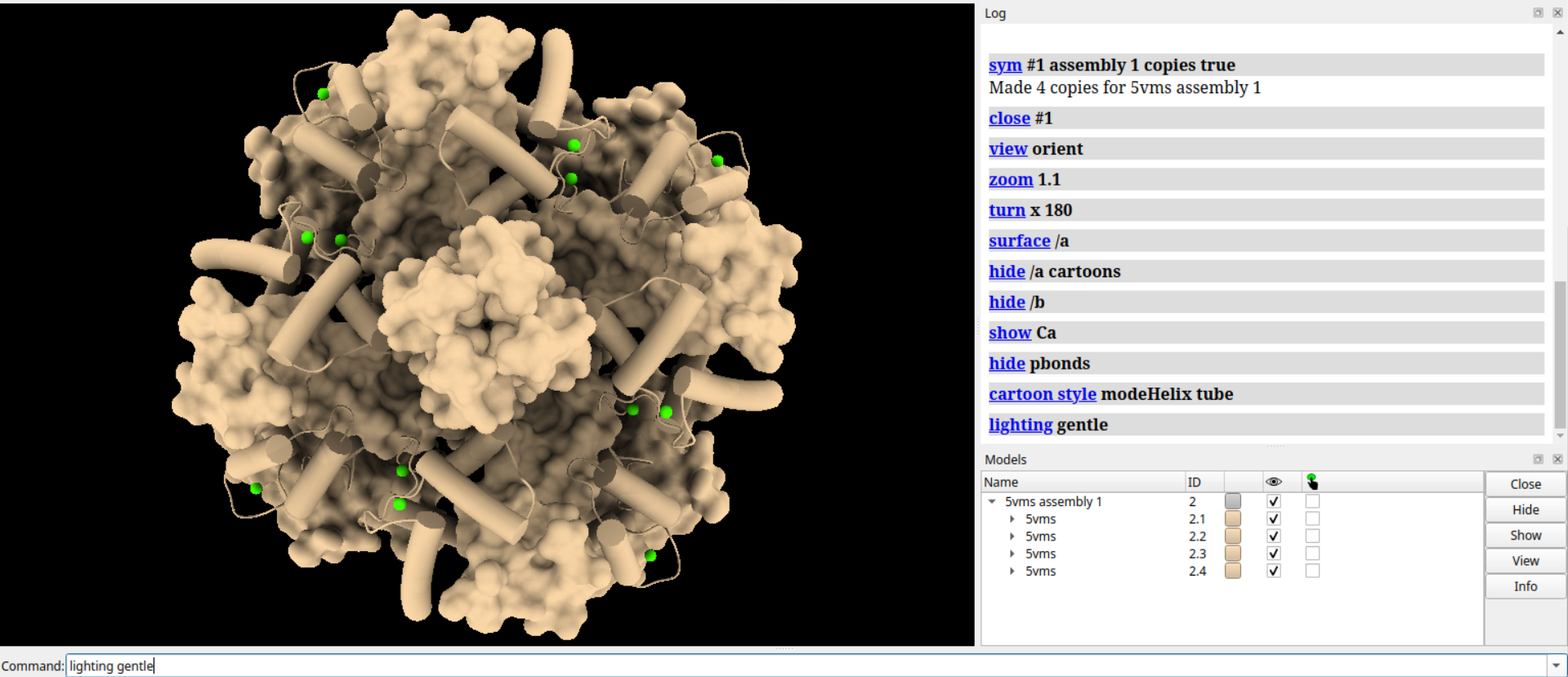
# 设置背景颜色为透明白色
set bgColor #ffffff00
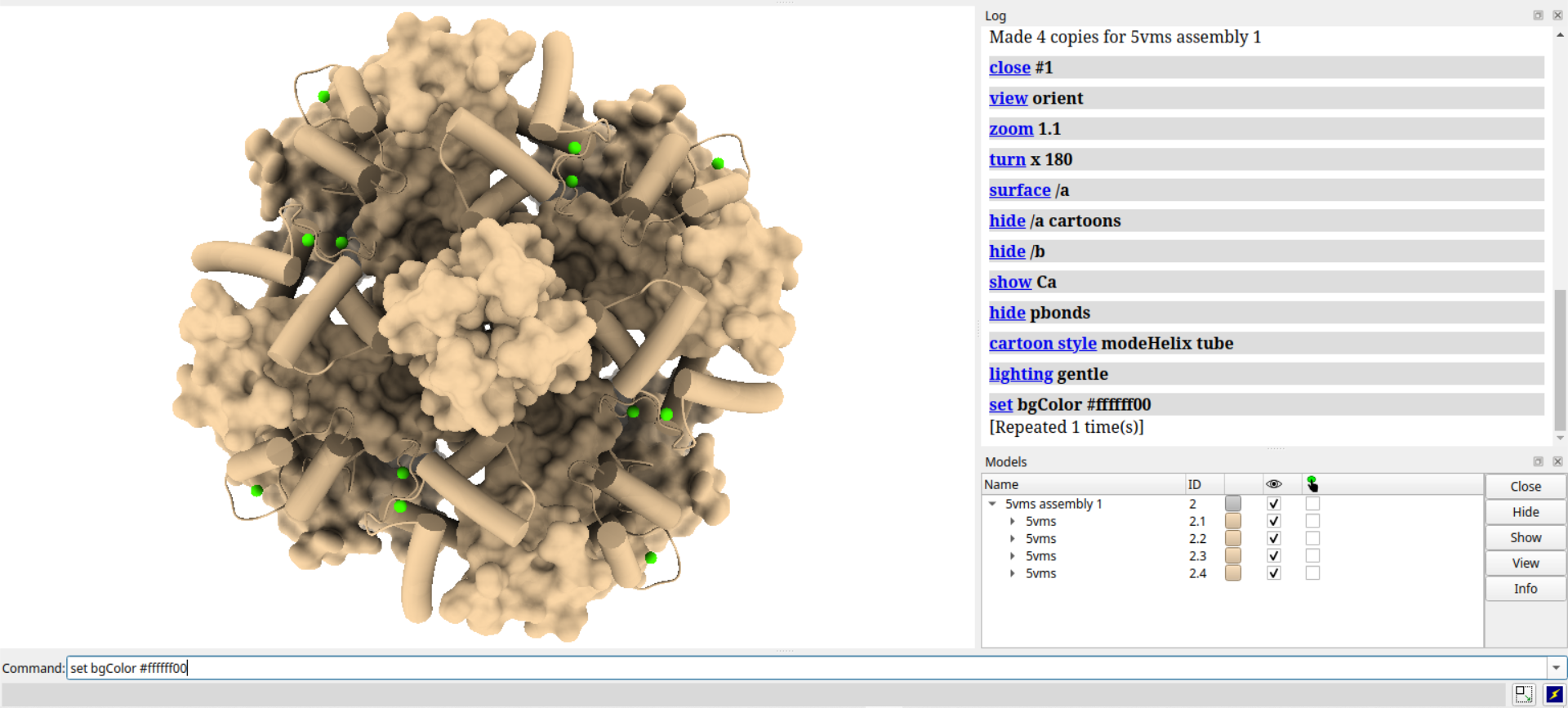
# 启用轮廓线
set silhouettes true
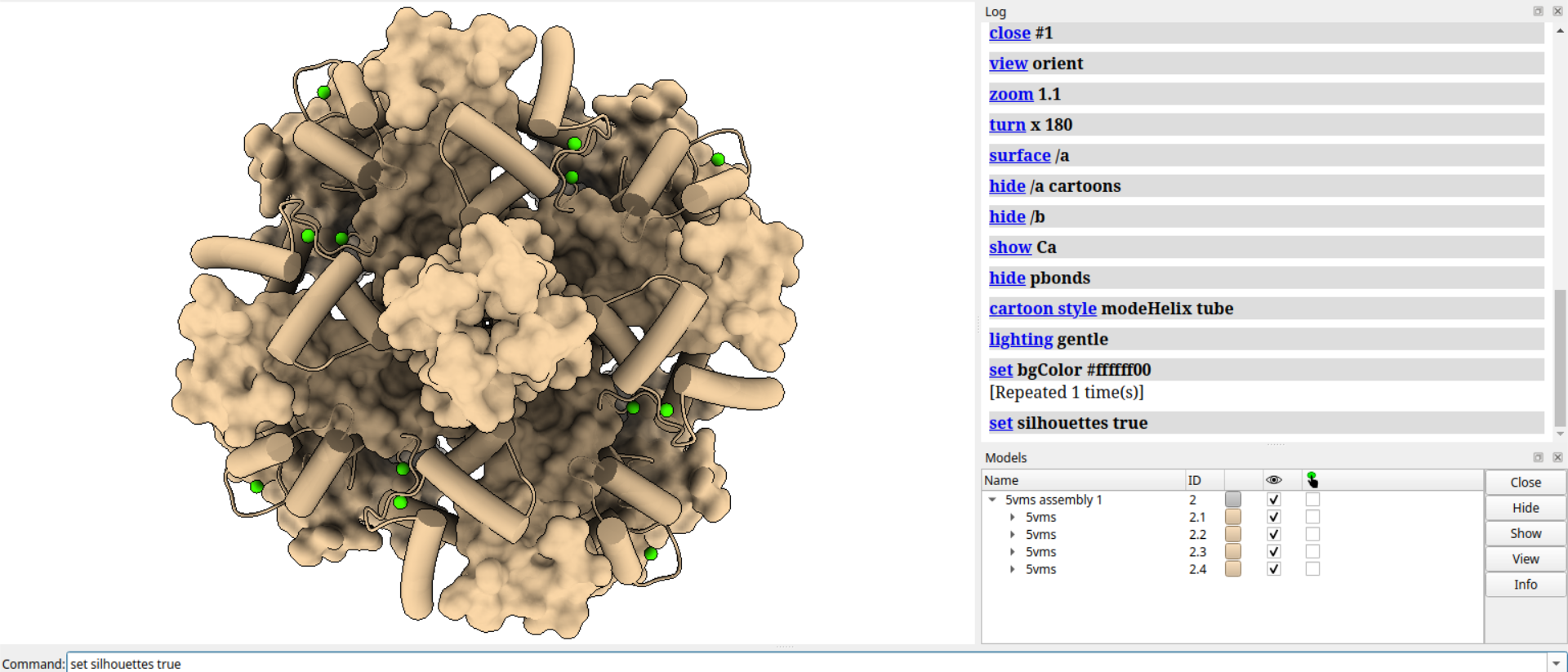
# 设置轮廓线深度为0.02
set silhouettedepth 0.02
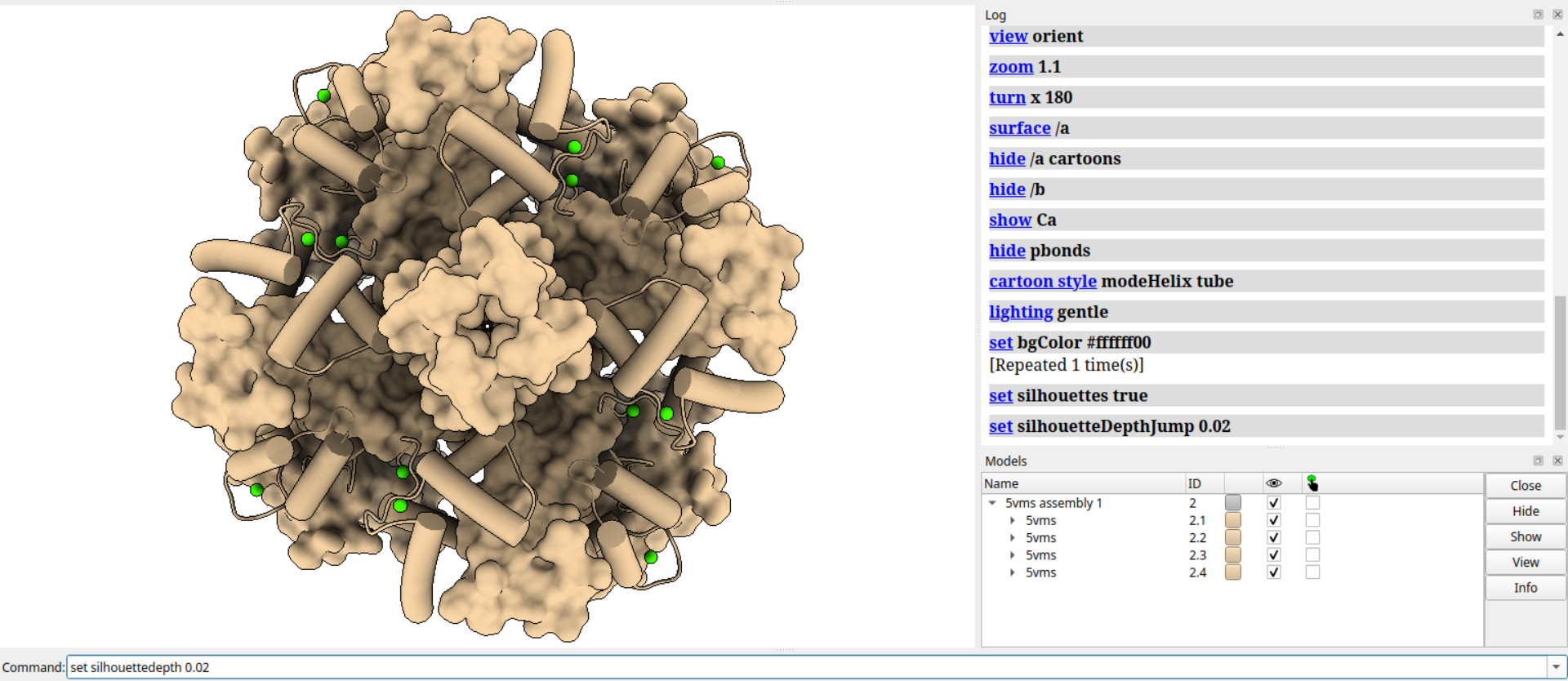
# 设置离子的原子大小为1.6
size ions atom 1.6
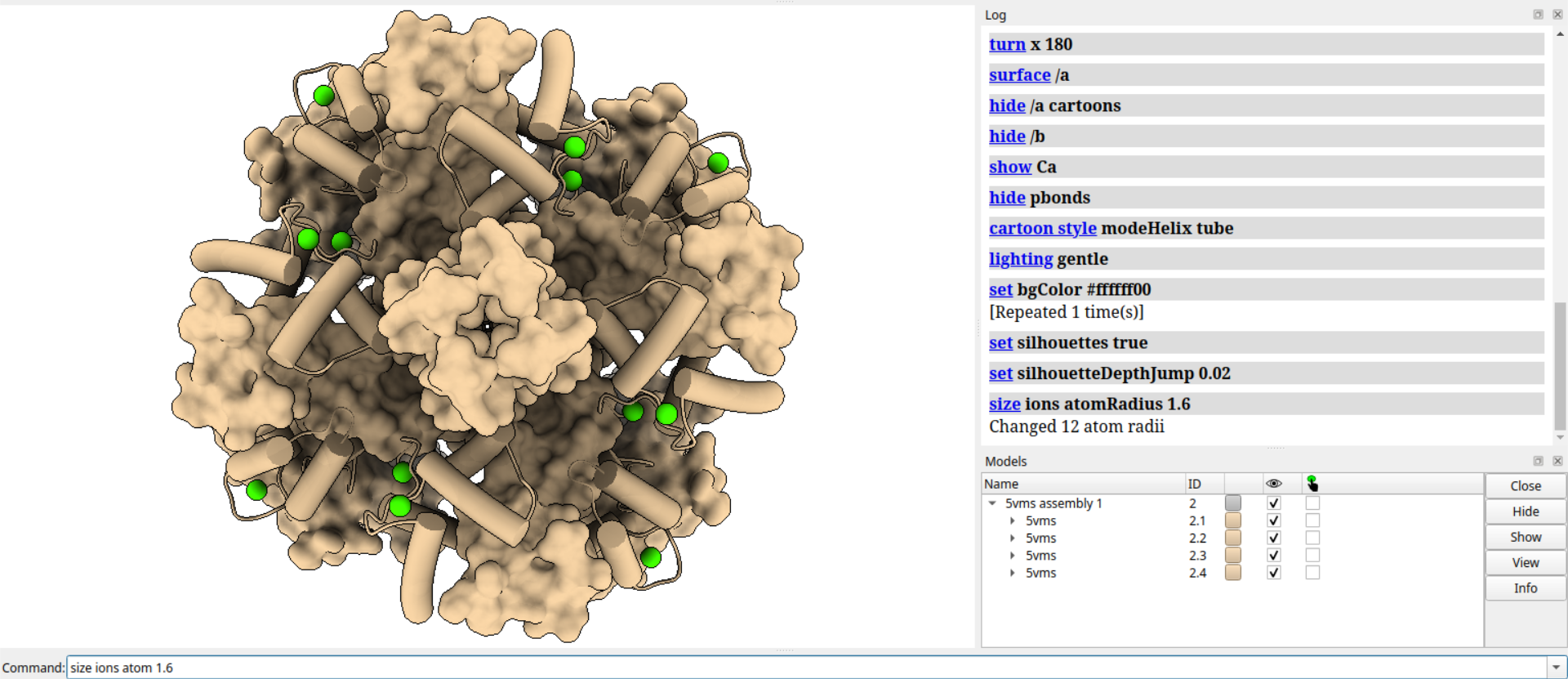
# 修改离子的颜色,色调增加45度
color modify ions hue +45

# 使用pubu-5调色板对彩虹颜色进行设置
rainbow palette pubu-5

# 使用pubu-5调色板对聚合物进行彩虹颜色设置
rainbow polymers palette pubu-5
2,如何使用chimeraX的动画命令来创建简单的分子旋转动画
open 3lel
show cartoons
hide atoms
turn x 90
movie record
turn y 2 180
wait 180
movie encode C:/path/test1.mp4
roll
stop
movie record
roll y 2 180
movie encode C:/path/test2.mp4
#open 3lel: 打开名为3lel的模型文件
#show cartoons: 显示模型的卡通表示
#hide atoms: 隐藏模型的原子表示
#turn x 90: 沿着X轴旋转90度
#movie record: 开始录制电影
#turn y 2 180: 沿着Y轴每次旋转2度,总共旋转180度
#wait 180: 等待180帧
#movie encode C:/path/test1.mp4: 将录制的动画编码保存为test1.mp4文件
#roll: 持续旋转模型
#stop: 停止当前的操作
#movie record: 重新开始录制电影
#roll y 2 180: 沿着Y轴每次旋转2度,总共旋转180度
#movie encode C:/path/test2.mp4: 将录制的动画编码保存为test2.mp4文件
在linux上操作,可以使用
open 3lel
show cartoons
hide atoms
turn x 90
movie record
turn y 2 180
wait 180
movie encode /mnt/sdb/zht/project/uniprot/pymol_chimerax/3lel_test1.mp4
roll
stop
movie record
roll y 2 180
movie encode /mnt/sdb/zht/project/uniprot/pymol_chimerax/3lel_test2.mp4
第一个test的视频动图如下(忘了在背景中去除前面的5vms了,将就看)——》无法上传算了

3,如何使用chimeraX的动画命令来创建和播放分子的动态效果,包括旋转、摇摆、变形和淡入淡出等?
open 3jc8
set bgColor white
rainbow polymers palette pubugn
view name p1
hide ~/Ba-Cb
view
view name p2
marker #2 position -165.2, 127.9, -152.2
show all
move y -90
view name p3
zoom 0.75
move y -120
view name p4
turn x 90
zoom 0.6
move y 120
view name p5
turn x -90
turn y 180
move y -100
view name p6
turn x -90
move y 105
view name p7
turn x 90
view
view name p8
view p8
movie record
fly p1 p2
crossfade
hide ~/Ba-Cb
roll y 4 90 center #2
crossfade
show ~/Ba-Cb
fly p2 60 p3 60 p4 p5 p6 p7
roll z 3 120 center #2 models /Ba-Cb
fly p7 p8
movie encode C:/path/test3.mp4
#open 3jc8: 打开名为3jc8的模型文件
#set bgColor white: 设置背景颜色为白色
#rainbow polymers palette pubugn: 为多聚物应用彩虹色彩方案pubugn
#view name p1: 将当前视图命名为p1
#hide ~/Ba-Cb: 隐藏名字中包含Ba-Cb的对象
#view: 保存当前视图
#view name p2: 将当前视图命名为p2
#marker #2 position -165.2, 127.9, -152.2: 在指定位置创建标记#2
#show all: 显示所有对象
#move y -90: 沿着Y轴移动-90单位
#view name p3: 将当前视图命名为p3
#zoom 0.75: 缩放视图至75%
#move y -120: 沿着Y轴移动-120单位
#view name p4: 将当前视图命名为p4
#turn x 90: 沿着X轴旋转90度
#zoom 0.6: 缩放视图至60%
#move y 120: 沿着Y轴移动120单位
#view name p5: 将当前视图命名为p5
#turn x -90: 沿着X轴旋转-90度
#turn y 180: 沿着Y轴旋转180度
#move y -100: 沿着Y轴移动-100单位
#view name p6: 将当前视图命名为p6
#turn x -90: 沿着X轴旋转-90度
#move y 105: 沿着Y轴移动105单位
#view name p7: 将当前视图命名为p7
#turn x 90: 沿着X轴旋转90度
#view: 保存当前视图
#view name p8: 将当前视图命名为p8
#view p8: 切换至视图p8
#movie record: 开始录制电影
#fly p1 p2: 在视图p1和p2之间创建飞行动画
#crossfade: 创建交叉淡出效果
#hide ~/Ba-Cb: 隐藏名字中包含Ba-Cb的对象
#roll y 4 90 center #2: 沿着Y轴旋转90度,中心是标记#2
#crossfade: 创建交叉淡出效果
#show ~/Ba-Cb: 显示名字中包含Ba-Cb的对象
#fly p2 60 p3 60 p4 p5 p6 p7: 创建一个复杂的飞行动画,从p2到p7的不同视图之间飞行
#roll z 3 120 center #2 models /Ba-Cb: 沿着Z轴旋转120度,中心是标记#2,只应用于/Ba-Cb模型
#fly p7 p8: 在视图p7和p8之间创建飞行动画
#movie encode C:/path/test3.mp4: 将录制的动画编码保存为test3.mp4文件
参数还是要自己相应设置:
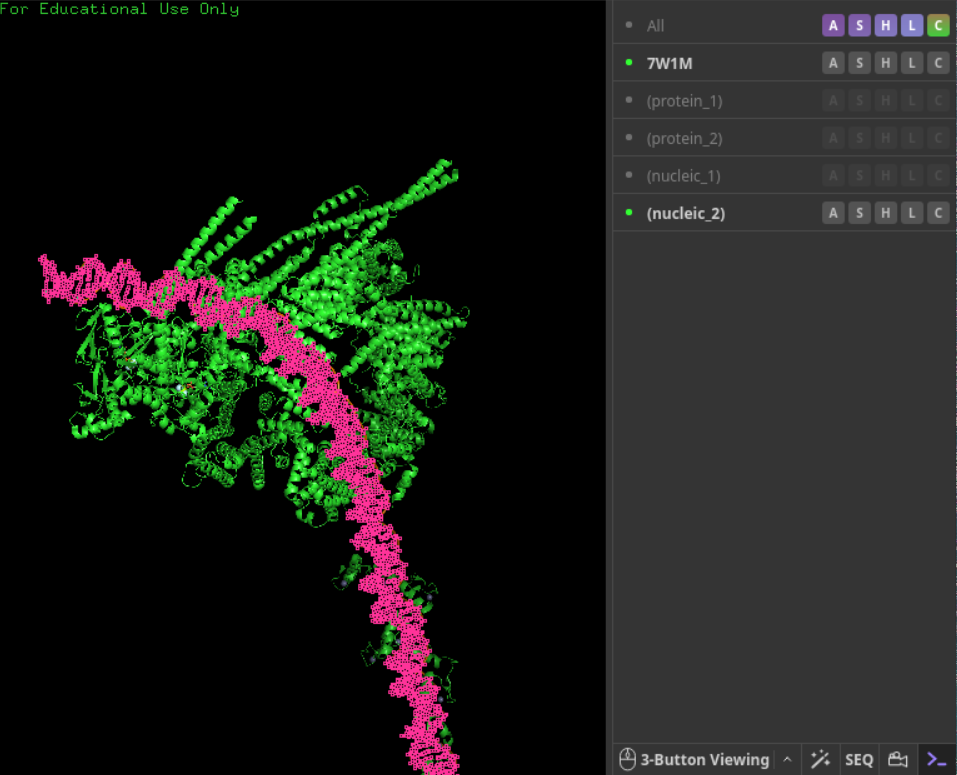
如果是7w1m效果就很差:
open 7W1M
set bgColor white
rainbow polymers palette pubugn
view name p1
hide ~/Ba-Cb
view
view name p2
marker #2 position -165.2, 127.9, -152.2
show all
move y -90
view name p3
zoom 0.75
move y -120
view name p4
turn x 90
zoom 0.6
move y 120
view name p5
turn x -90
turn y 180
move y -100
view name p6
turn x -90
move y 105
view name p7
turn x 90
view
view name p8
view p8
movie record
fly p1 p2
crossfade
hide ~/Ba-Cb
roll y 4 90 center #2
crossfade
show ~/Ba-Cb
fly p2 60 p3 60 p4 p5 p6 p7
roll z 3 120 center #2 models /Ba-Cb
fly p7 p8
movie encode /mnt/sdb/zht/project/uniprot/pymol_chimerax/7w1m_1.mp4

















 6586
6586

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









