






我们前面介绍了 如何通过AlphaFold在线版对蛋白结构进行预测,但预测仅仅是一个开始,对于预测得到的PDB文件我们还需要选择Pymol、Chimera等软件对结果进行可视化。Pymol需要大量的代码来得到我们想要的结果,上手难度较高;而今天我们介绍的ChimeraX是一款简单易上手的软件,本次主要介绍ChimeraX软件和蛋白结构的导入。
01: ChimeraX 介绍
UCSF ChimeraX(简称 ChimeraX )是一款由美国加州大学开发的免费分子可视化工具,目前已经更新到1.5版本(点击阅读原文跳转下载)。该软件下载速度慢,需要科学上网(VPN),后台回复ChimeraX可得到软件安装包。ChimeraX 可以从PDB数据库中直接导入蛋白结构,也可以将AlphaFold预测的结果进行可视化,该软件能展示蛋白三级结构的形状、蛋白之间互作界面预测的结果。软件的安装比较简单,本文不再赘述,着重介绍模型选择和简单可视化操作。
02: PDB 文件的选择
AlphaFold预测结束后我们会得到一个 .zip 格式的压缩包,解压缩打开后主要有 .JSON 、 .PDB 、.PNG 和文本文档类格式文件,我们需要的是 .PDB 文件,这里面存放着我们预测的蛋白结构结果,如果我们对一个蛋白预测了好几个模型,那么我们该如何选择哪个模型进行可视化?因此我们还要关注 .PNG 格式文件和文本文档格式中的settings文件,因为通过这些文件可以判断我们蛋白结构预测的质量高低。当然为了我们后续可以更好的实验,建议最好选择所有预测的模型来进行可视化分析,毕竟AlphaFold只是预测出来的结果,其可信度仍有待验证。

.PDB格式文件是我们需要在ChimeraX中打开的文件;.PNG格式文件反馈蛋白结构预测质量;settings文件反馈蛋白结构预测质量
打开所有 .PNG 格式文件如下图,每一张图都是对我们预测的蛋白模型的精准度反映。我们主要看图⑤,可以清楚的看到蛋白不同区段的预测精准度,峰值越高则越精准,通过该图我们选择预测结果最精准的PDB文件进行可视化,在此选rank1用于后续的分析。(如果我们想看结构域之间的互作,可直接在AlphaFold预测时只输入结构域蛋白序列,小编认为这样预测更可信)

我们还可以通过settings文件看到预测时的参数pLDDT和pTMscorc值,结果也表明rank1为可信度高的模型。

03:使用 ChimeraX 可视化蛋白预测结果
打开 ChimeraX 后依次点击File、Open选择我们要打开的PDB文件,就可以看到我们预测的蛋白模型。
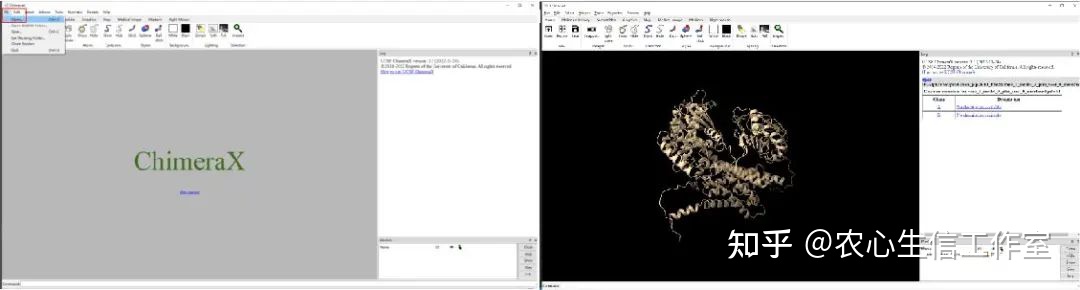
下面我们简单介绍一下这个界面包含的信息:
图中1位置是我们常用到的可视化指示命令工具,可满足对蛋白结构进行各种操作。当然如果个人对代码比较了解可以直接在最下面的Command中输入命令来执行操作。
图中2位置是蛋白可视化的界面,用于查看蛋白结构。使用鼠标滚轮可以进行蛋白缩放,使用左键可以选择和转动模型,使用右键可以移动模型。
图中3位置会实时显示所执行的命令以及执行完命令后的结果反馈,例如想选中某一条蛋白链可以在Chain中直接选择,后在此界面可以看到该蛋白链的数据。
图中4位置是所打开的Models目录,如果打开多个PDB文件这里就会显示每个文件,我们选择需要的Model进行观察。

关于 ChimeraX 具体怎么使用我们很快会给大家介绍,如果大家有什么问题或者想要了解的内容,欢迎关注同名公众号。















 2540
2540











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









