






作者:青萍,你好
出处:https://www.cnblogs.com/timeisbiggestboss/p/8856888.html
1.supplementary alignment
supplementary alignment是指一条read的一部分和参考区域1比对成功,另一部分和参考区域2比对成功,参考区域1和参考区域2没有交集(或很少),那么一条read就会产生两条sam文件,
将其中的一条sam文件作为represent alignment,而另一条作为supplementary alignment (flag为2048)。
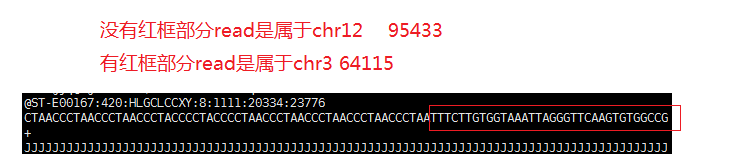
将上面的fastq文件去跑bwa,read有两条sam文件,第二条的flag值为2048:

2.bwa mem的-M -Y参数:
-M:mark shorter split hits as secondary。就是把supplemenary alignment 变为no primary(flag值256) 。下面是bwa mem -M的结果

-Y:use soft clipping for supplementary alignments。把默认的hard clip变为soft clip。hard clip 不会显示不匹配的碱基串,soft clip会显示不匹配的碱基串。下面是bwa mem -Y的结果(58H34M变为58S34M)

3.secondary(no primary)是指这条read在基因组上有多个匹配区域(>=2),可以是read的同一部分有不同匹配区域,也可以是一条read上的不同区域。所以supplemenary aligment应该算是secondary的子集。
许多处理bam的软件不会去处理supplemenary(split alignments),比如Picard’s markDuplicates,所以可能需要用-M把supplemenary 转换为secondary。
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
生信分析
我们能够承接所有 NGS 组学数据分析业务,包括但不限于 WGS / WES / RNA-seq 等。基因组组装、注释,以及各种重测序业务都可以与简说基因合作。
生信培训
简说基因的生信培训班,荣获学员的一致好评。如果你也对生物信息学感兴趣,欢迎来跟简说基因,学真生信。
联系方式
QQ交流群(免费):925694514
微信交流群(免费):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy














 1553
1553











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









