







引言
参考基因组是现代遗传学的基石,更是生物信息学研究的基础。不论你研究的是DNA、RNA,还是蛋白,你都无法避开参考基因组而进行你的研究。在生物信息分析过程中,参考基因组最常用的一个场景就是序列比对---将测序的read比对回参考基因组上。那么,如果我们想知道参考基因组某个位置或者某个区域的碱基序列,我们应该如何快速的解决这个问题呢?本文将介绍三种解决方案。
参考基因组格式
参考基因组通常以FASTA格式存在。在生物信息学中,FASTA格式是一种用于记录核酸序列或肽序列的文本格式,其中的核酸或氨基酸均以单个字母编码呈现。该格式同时还允许在序列之前定义名称和编写注释。这一格式最初由FASTA软件包定义,但现今已是生物信息学领域的一项标准。(维基百科)
FASTA文件中存储的一条完整的序列,由两部分组成:
1. 开头的单行描述行
描述行以“>”开头,用以和数据行分开,后紧接的内容为该序列的标识符,该行剩余部分则为序列的描述(标识符与描述均非必须)
2. 紧随其后的序列行
序列行可以是多行,序列的结束以下一条序列的“>”出现为标识。

FASTA格式示例
提取位置碱基
接下来我们以chr5:8397384-8397384为例,分别介绍三种方法提取某个位置的碱基。
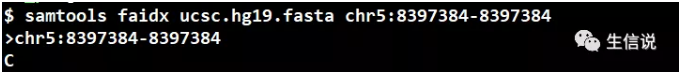
01 利用samtools faidx
samtools faidx常常用来对参考基因组建立索引,但它还有个鲜为人知的功能,就是序列提取,如下:
02 利用bedtools getfasta
bedtools说明文档中对getfasta的描述是“Extract DNA sequences into a fasta file based on feature coordinates.”显而易见,bedtools getfasta的功能就是根据坐标信息提取序列信息。操作如下:
![]()
bedtools getfasta有三个必选参数:-fi即参考基因组fasta文件;-bed即需要提取的位置坐标信息,格式:chr\tstart\tend;-fo:输出文件。
有一点需要说明,bedtools接收的是bed文件,而bed文件时0-based,因此我们要提取chr5:8397384-8397384位置的碱基,输入文件pos.bed中需要写入:chr5\t8397383\t8397384

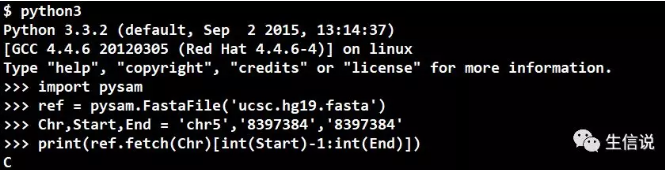
03 利用pysam模块
直接上代码吧!
性能比较
三种方法耗时如下:
| samtools | bedtools | python | |
| time | 0.002s | 0.034s | 2.077s |
可以看出,samtools和bedtools的性能很好,python的性能就比较尴尬了。其实,个人比较推荐bedtools,比较容易进行批量处理,把想处理的位置信息写到输出文件,然后就可以轻松的进行序列提取。















 1797
1797











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









