






宏基因组组装的基因组目录揭示了马肠道耐药基因特征和运动能力相关的微生物
Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse

Research,2023-1-12,Microbiome, [IF 16.837]
DOI:https://doi.org/10.1186/s40168-022-01448-z
第一作者:Cunyuan Li (李村院);Xiaoyue Li (李晓悦)
通讯作者:Shengwei Hu (胡圣伟)
主要单位:石河子大学生命科学学院 (College of Life Science, Shihezi University, Shihezi, 832003, Xinjiang, China)
- 摘要 -
背景:作为一种对人类至关重要的驯化物种,马作为运动、休闲、食品生产和运输工具的来源在世界范围内饲养。肠道微生物群在马的健康、疾病、运动表现和行为中起着重要作用。
结果:在这里,使用来自242匹马的肠道样本的约2.2 Tb宏基因组测序数据,包括来自盲肠的110个样本和来自直肠(粪便)的132个样本,我们组装了4142个微生物宏基因组组装基因组(MAG),其中4015个(96.93%)似乎对应于新物种。从长读数数据中,我们成功组装了代表新物种的13个环状全染色体细菌基因组。MAG包含超过313,568种预测的碳水化合物活性酶(CAZy),其中超过59.77%在CAZy公共数据库中具有低相似性匹配。在MAG中发现了高丰度和多样性的抗生素耐药性基因(ARG),可能表明抗生素在马的管理中被广泛使用。赛马中至少36 个MAG的丰度(例如属于Lachnospiraceae,Oscillospiraceae和Ruminococcus的MAG)在赛马中高于非赛马。这些赛马中富含的MAG含有通过纤维发酵生产乙酸盐和丁酸盐的主要途径中的每个基因,具有更多短链脂肪酸可用于促进运动表现的潜力。
结论:总体而言,我们从马肠道中的短读和长读序列数据中组装了4142 MAG。我们的数据集代表了马肠道微生物组的详尽微生物基因组目录,并为发现运动能力相关的微生物和研究马肠道微生物组提供了宝贵的资源。
关键词:运动能力,耐药基因,肠道,马,宏基因组组装基因组,Nanopore测序
- 引言 -
马(Equus caballus)在世界范围内用于休闲,赛马,运输,农业和林业活动,并且在发展中国家广泛用于生产奶制品和动物蛋白。全球估计有5900万匹马,每年的经济影响约为3000亿美元。因此,详细了解马的生物学以潜在地改善马在人类活动中的使用是非常重要的。
马已被用作是研究后肠发酵食草动物消化的模型系统,包括驴,斑马,犀牛和大象,以及研究反刍动物的盲肠消化。马的肠道含有大量的共生细菌,真菌,古细菌,原生动物和病毒,它们为动物提供水解酶,通过纤维发酵将碳水化合物转化为能量。这些微生物群在马体内产生的代谢物在宿主健康、疾病、发育甚至行为中起着重要作用。先前的研究报告表明,肠道微生物群与马疾病(例如结肠炎、蹄叶炎、牧草病、哮喘、腹泻)和运动有关。Mach等人使用血液转录组、血液代谢组和粪便微生物组来研究马匹赛前赛后的耐力,并提出肠-线粒体轴与运动表现有关。因此,深入了解马肠道的微生物组成,通过饮食干预或添加益生菌来操纵微生物群,为提高运动表现和改善动物健康的维持提供了机会。
马肠道中的微生物群组成主要基于16S扩增子测序进行了部分表征,最近,已经报道了马肠道微生物组的基因目录和数百个MAG,但仍然缺乏对马微生物群组成和功能的大规模研究。高通量测序和宏基因组分箱技术的出现使得大规模获得近乎完整的宏基因组组装基因组(MAG)成为可能。宏基因组测序技术可以在肠道微生物中鉴定出大量以前未知的细菌种类,并已用于在基因组水平上表征这些微生物的功能。这项技术已经从人类,反刍动物,鸡,猪和马中产生了数千个MAG。此外,长读长测序可以通过增加基因组组装的连续性来提高组装质量。因此,使用高通量测序技术与长读长测序相结合,为表征马微生物组的微生物组成和功能提供了一种有希望的策略。此外,马肠道中增强性能的微生物和新型酶(例如CAZy)可能会通过宏基因组学被深入探索。
在这项研究中,我们旨在开发一种微生物基因组资源,用于研究马肠道微生物组,并使用该资源回答有关肠道微生物群与赛马表现如何相关的问题。我们使用了来自中国两个省份的242匹马的肠道样本。进行了大规模的宏基因组测序扫描,以表征马肠道中的微生物群组成。此外,我们揭示了马的耐药性特征和运动表现相关微生物。这项研究提供了马体内MAG的详尽目录,并回答了有关肠道微生物组与马性能之间关系的重要问题。
- 结果与讨论 -
样本和宏基因组测序数据
Samples and metagenomic sequencing data
为了提供研究马肠道微生物组的资源,对来自242匹不同年龄(范围:1-11岁)、性别(雌性、雄性)和品种(纯种马、伊犁马、伊犁赛马、藏马或设特兰矮种马)的110个盲肠和132个直肠(粪便)样本进行了宏基因组测序,这些样本保持在不同的饮食下(图1A)。使用高通量测序,我们从242个样品中获得了2.267 Tb Illumina测序数据。经过质控,共保留了2.264 Tb的干净、高质量的数据,有效数据质控率为99.86%。为了评估可以从这些样本中鉴定出的基因总数,随机抽样100次,进行了稀疏分析。稀疏曲线接近饱和,表明序列数据足以对马肠道微生物群进行基因组分析,并且很少有新基因未被发现。此外,为了提高数据组装的质量,我们利用Nanopore测序对两个样品(HGM35和HGM77)进行了测序,获得了0.057 Tb的长读长测序数据。
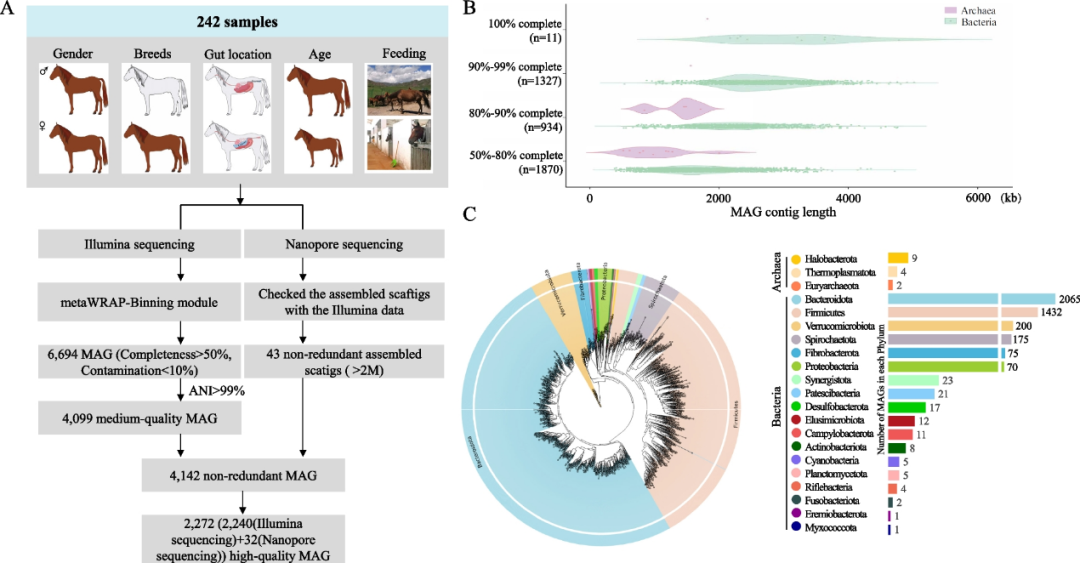
图1 MAG构建流程及组装的MAG的基本信息。
(A) 242个马的基本信息和使用Illumina和Nanopore技术从242个马宏基因组数据集构建MAG流程示意图。(B) 基因组完整性的分布和MAG按质量等级的分类。(C)来自马肠道的4142个 MAG的系统发育树
来自马肠道的4142个微生物基因组的组装
Assembly of 4142 microbial genomes from the horse gut
我们使用metaWRAP-Binning模块生成了6408个bins(MaxBin2),7495个bins(metaBAT2)和12,415个bins(CONCOCT)(图1A,左侧)。经过去重复(平均核苷酸同一性(ANI)≤99%)和质量评估,我们获得了最终的4099个MAG,达到或超过先前建立的中等质量标准(完整性≥50%,污染≤10%)。通过Nanopore测序方法从两个测序样品(HGM35 和 HGM77)中生成了另外43个基因组(图 1A,右侧)。因此,我们使用Illumina和Nanopore测序技术总共组装了4142个MAG。在4142个MAG中,2272个是高质量基因组(定义为完整性>80%和污染率<10%)(图1B);646个完整性>95%且污染率< 5%;46个完整性>97%且污染为0%。
为了对4142个MAG进行分类,将这些MAG序列与基因组分类数据库(GTDB)比对。进一步分析表明,4142个MAG中有126个在种水平上被鉴定出来,3462个在属水平上被鉴定出来,4127个在科水平上被鉴定出来,4139个在目水平上被鉴定出来,4142个在纲水平上被鉴定出来。在菌株水平和物种水平基因组bins阈值的ANI分别为99%和95%的MAG聚类,我们发现3253个MAG可以被认为是物种水平的基因组。所有4142个MAG均被分类为GTDB预测的分类群(图1C),包括18个细菌门(n = 4127 MAG)和3个古菌门(n = 15 MAG)。如图1C所示,前10个细菌门是Bacteroidota (2065个MAG)、Firmicutes (1432个MAG)、Verrucomicrobiota(200个 MAG)、Spirochaetota(175个MAG)、Fibrobacterota(75个MAG)、Proteobacteria(70个MAG)、Synergistota(23个MAG)、Patescibacteria(64个MAG)、Desulfobacterota(17个MAG)、Elusimicrobiota(12个MAG)和Campylobacterota(11个MAG)。4142个MAG中的4015个(>96.93%)与GTDB中的参考基因组不匹配,因此代表了本研究中首次鉴定的未知物种或菌株。通过对先前研究中的马MAG基于95% ANI水平进行去重,我们发现3936个MAG在这项研究中是唯一的。通过比较242匹马中鉴定的MAG,我们发现至少90%的样本中存在36个核心MAG,包括Bacteroidota(MAG = 16),Firmicutes(MAG = 18)和Verrucomicrobiota(MAG = 2),这些门也存在于报道的马肠道宏基因组数据中。同时,在90%的样品中相对丰度<1%的MAG被认为是稀有的,共有43个MAG被确定为稀有。
在这项研究获得的15个古菌MAG中,有13个属于未知物种。所有古菌MAG被分配到三个门:Halobacterota (n = 9),Thermoplasmatota (n = 4)和Euryarchaeota (n = 2)。为了揭示古菌MAG的潜在功能,基于完整性>80%的阈值选择了6个高质量的古菌MAG,并进一步分析了甲烷生成途径基因的存在。所有6个高质量古菌MAG都含有>500个产甲烷基因。有趣的是,MAG23.bin.19(完整性为100%且污染率为0%)包含这些古菌MAG中鉴定出的产甲烷基因最多,可以利用所有三种已知途径(氢营养型,乙酸分解型和甲基营养途径)产生甲烷。MAG23.bin.19被分配到Methanobrevibacter smithii。先前的研究报道,Methanobrevibacter smithii是人类肠道中的主要古细菌(占所有厌氧菌的10%),并促进了人类大肠中甲烷的产生。同时,Methanobrevibacter smithii也是瘤胃中重要的产甲烷菌。总之,我们的结果表明,MAG23.bin.19可能是马甲烷产生的主要贡献者。Methanobrevibacter smithii的完整基因组信息可能为减少甲烷产生提供新的靶点,尽管未来需要进一步的实验分析来证实这一点。
从长读数据中组装13个未知物种的第一个完整的环状基因组
Assembling the first complete, circularized genomes of 13 unknown species from long-read data
为了生成完整的微生物基因组,对两个宏基因组样本(HGM35,母;HGM77,公)进行了Nanopore测序,产生了超过50.13 Gb的clean data(超过57.71 Gb的raw data,有效率>86.85%)。平均read长度为55,063 bp,优于先前研究报告的数据。在组装这些long reads后,我们得到了一个长度为739 Mb的序列,N50值达到217kb。如图所示(图 1A),我们组装了32个新的高质量 MAG,这些MAG以前在公共数据库中没有被识别出来。与Illumina数据相比,我们观察到Nanopore测序数据在组装长度和contig长度方面具有优势(图2A)。
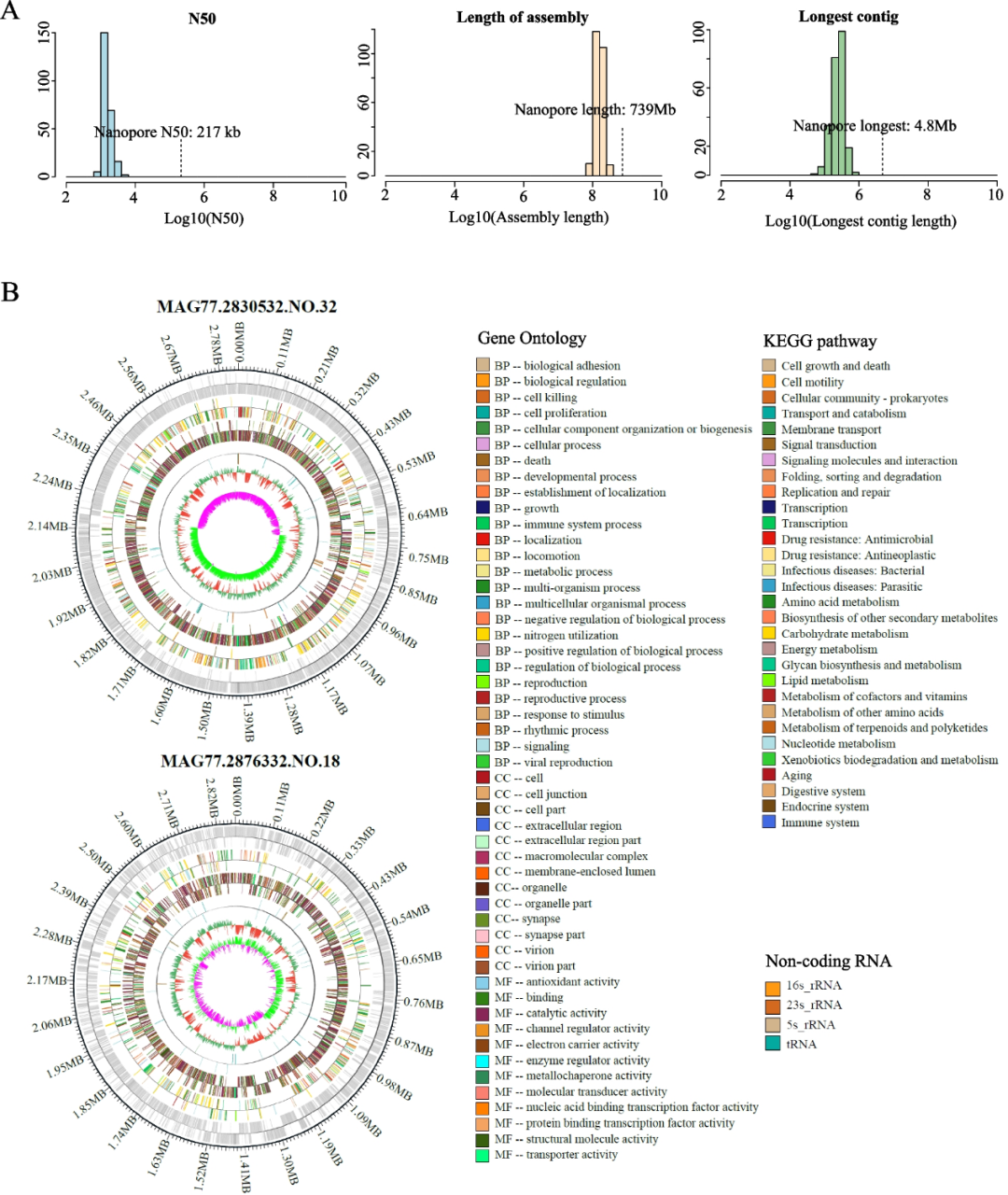
图2 从长读长数据组装第一个完整的环状基因组。
(A) Illumina和Nanopore序列组装结果的三个重要指标的统计分布。(B) 2个环状基因组概述。
long reads可用于从单一scaffold组装近乎完整的环状MAG(cMAG),这大大提高了装配的精度。在long reads生成的32个MAG中,有13个成功组装成cMAG,这是长读长组装的重要成果。这些cMAG的平均完整性和污染率分别为93.25%和小于0.99%。累积GC偏倚和基因组GC含量用于验证cMAG的组装质量。
将这13个cMAG与NR数据库和基因组分类数据库工具包(GTDB-Tk)进一步进行比较,以鉴定同源物种。在这些同源物种中,cMAG的ANI最高为83.69%。这种相对较低的ANI表明这些cMAG的新颖性,并代表了各自物种的第一个环状的完整基因组。这些cMAG包含完整的细菌基因组信息,包括16S,5S和23S rRNA操纵子和tRNA操纵子的多个拷贝。接下来,我们确定了最高质量的组装基因组,基于污染率进行进一步分析,并选择了MAG77.2830532.NO.32,MAG77.2876332.NO.18和MAG77.3034135.NO.12,所有这些基因组的污染评分均为0%(图2B)。与公共数据库的比较确定MAG77.2830532.NO.32为Anaerovibrio lipolyticus(ANI < 73.90%),MAG77.2876332.NO.18为Ruminococcaceae细菌的新物种(ANI < 71.20%)(图2B),MAG77.3034135.NO.12被确定为未知Ruminococcus sp.(ANI<79.17%)。我们在这些基因组中检索了全长16S序列,通过与NCBI数据库进行比较发现,匹配率最高的是未培养的瘤胃Anaerovibrio sp(97.30%同一性)、未培养的Ruminococcus sp.(95.60%同一性)和未培养的Ruminococcaceae细菌(97.30%同一性)。根据基于16S rRNA的分类学的一般标准,只有MAG77.2876332.NO.18可以被认为是一个新物种,这进一步表明了微生物分类群基因组测序的准确性。这些结果表明,MAG的长读长组装不仅增加了基因组组装的完整性,而且还揭示了以前未解决的基因组特征和分类信息。
数十万种新的CAZy
Hundreds of thousands of novel CAZy
接下来,我们通过搜索KEGG和CAZy数据库来分析马宏基因组的蛋白质组学内容和功能。2272个高质量MAG总共包含4,632,123个预测蛋白质。通过将本研究的基因目录与Mach等人,Youngblut等人和Gilroy等人报告的已发表的马数据进行比较,我们发现我们的数据大大扩展了马肠道微生物基因目录。在4,632,123种预测蛋白质中,其中6.77%(313,568)被预测至少具有一个CAZy功能。313,568种CAZy蛋白包括130,001种糖基水解酶(GH)、73,365 种糖基转移酶(GT)、52,961种碳水化合物结合模块(CBM)、46,437种羧酯酶(CE)、6320种多糖裂解酶(PL)和4484种具有辅助活性(AA)的蛋白质(图 3A)。这些蛋白质在我们鉴定的分类群的基因组中分布不均匀。例如,GH和GT尤其富集在Verrucomicrobiota和Firmicutes中(图3B)。我们进一步分析了预测的CAZymes与当前CAZy数据库的相似性。在313,568个CAZy蛋白中,只有126,139个(40.23%)显示出高度相似的匹配性,一致性≥95%,这表明我们预测的蛋白质中有187,429个是新的CAZy(图3C)。在预测的CAZymes的所有类别中,GH在公共数据库中与CAZy的氨基酸水平序列同一性最高(87.72%),而AA的一致性最低,仅为49.18%(图3C),表明公共数据库中缺少很大一部分CAZymes多样性。
我们将这项研究的CAZyme概况与Mach等人,Youngblut等人和Gilroy等人最近提供的先前马研究的概况进行了比较。我们发现我们的数据显著扩大了马肠道中CAZy的数量(图3D)。对于这六类CAZy,包括GT,GH,AA,PL,CE和CBM,我们的数据将马肠道中CAZy的数量至少增加了一个数量级,尽管我们的研究与先前研究之间的不一致与样本收集或测序方法有关,可能会影响这些数据集之间的比较。我们进一步分析了马的性别或品种对CAZy概况的影响。马肠道CAZy基因丰度与性别(R2=0.04,P=0.032)和品种(R2=0.2107,P=0.001)显著相关。
此外,我们在556个Bacteroides物种中检测到4492个多糖利用位点(PUL),它们可以降解动物消化系统中的各种碳水化合物底物。556个Bacteroides基因组中的大多数至少具有一个PUL,特别是Bacteroides fragilis(MAG117.bin.13),含有多达48个PUL。总的来说,这些新型CAZy和潜在的纤维素降解细菌的鉴定将有助于更好地了解马肠道中的碳水化合物代谢,并为发酵生物技术行业提供丰富的新型酶和微生物来源。
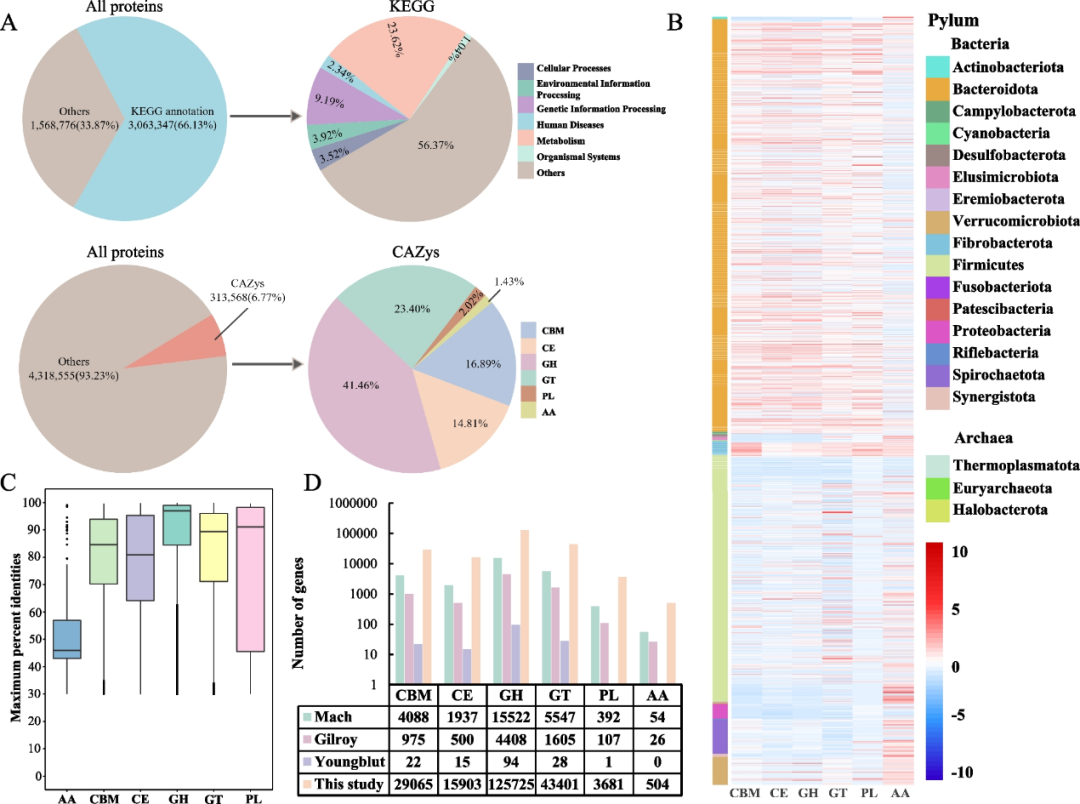
图3 马肠道中MAG的功能注释。
(A) 马微生物蛋白的功能注释。(B) CAZy分布的热图。(C) 本研究中CAZy与公共数据库的序列同一性。(D) CAZy基因数量与先前研究的比较。
马肠道作为抗生素抗性基因的储存库
The horse gut as a reservoir of antibiotic resistance genes
为了表征马肠道抗生素抗性基因(ARG),我们检测了242个马肠道样本中2272个高质量MAG中ARG的分布。我们在马肠道MAG中根据25个药物类别总共确定了266种独特类型的ARG(图4A)。ARG和药物类别的数量均少于猪和牛肠道中报道的数量,这可能与遗传,饮食和生活中接触抗生素有关。Firmicutes和Bacteroidota是ARG数量最多的细菌群,我们还预测了三个古菌类群的ARG,包括Euryarchaeota和Halobacterota(图4A)。氨基糖苷类、氨基香豆素和四环素ARG在马的肠道中普遍存在(图4A),这与先前在牛、羊、猪、鸡和马肠道微生物中的ARG研究一致。然而,氨基糖苷类抗性基因是马肠道中最丰富的ARG。鉴于氨基糖苷类药物被广泛用于治疗马的呼吸系统疾病、化脓性腹膜炎、急性发热性腹泻和马蜂窝组织炎,高丰度氨基糖苷类抗性基因对马的健康有潜在的负面影响。
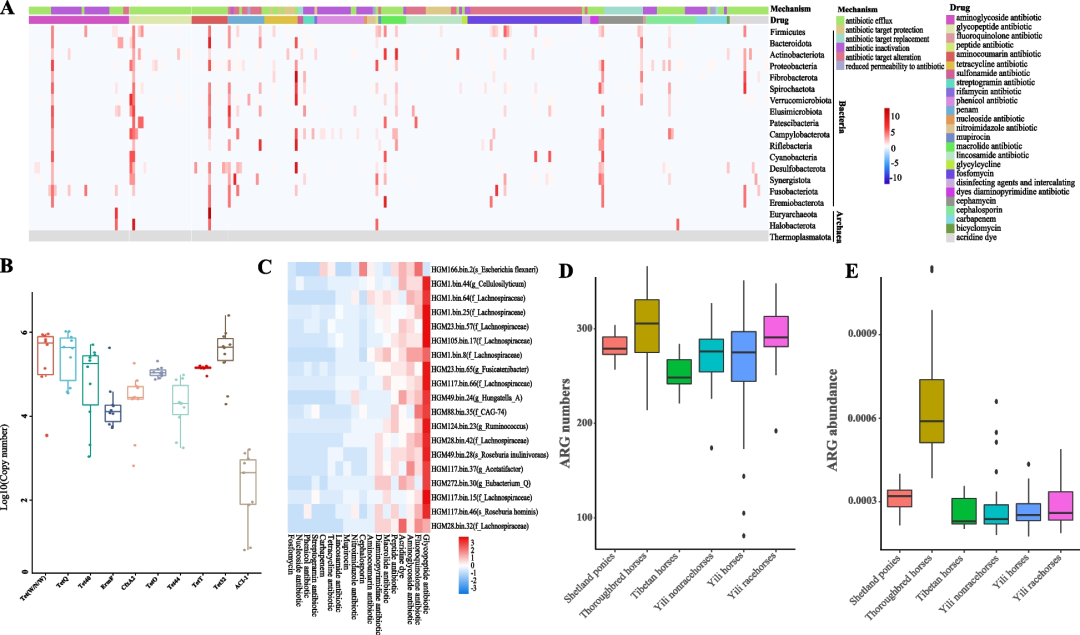
图4 马肠道作为抗生素抗性基因的储存库。
(A) ARG分布热图。(B) 随机选择的10个ARG的实时RT-PCR分析结果。(C) ARG的数量排名前20的MAG中抗性相关药物类别的热图。(D) 不同品种的马肠道微生物中ARG的基因数量。(E) 不同品种的马肠道微生物中ARG的相对丰度。
此外,共有2194个MAG(96.57%)含有五个或更多的ARG,表明ARG在马肠道微生物组中广泛存在。我们随机选择的10个ARG通过实时RT-PCR验证了这些ARG的转录活性,结果表明这些预测基因具有真正的抗性功能(图4B)。令人惊讶的是,发现Escherichia coli菌株(MAG166.bin.2)含有属于13个药物类别的82种独特类型的ARG(图4C)。该菌株含有多种针对氟喹诺酮类抗生素、头孢菌素、吖啶染料、碳青霉烯类、氨基糖苷类抗生素、肽类抗生素和畜牧生产中常用的四环素抗生素的抗性基因。考虑到E. coli的共同致病性,这一发现表明该菌株可能是一种潜在的超级耐药细菌。此外,我们在MAG49.bin.28(Roseburia)、MAG250.bin.22(Prevotella)和MAG77.2754209.NO.29(Akkermansia)中发现了大量的ARG,其中的成员被广泛报道具有益生菌作用。虽然抗生素抗性可用于促进这些菌株未来的分离和培养,但我们也可以考虑这些ARG可能对宿主肠道微生物组造成的潜在危害。
然后,对六个品种(伊犁马、纯种马、设特兰小马、藏马、伊犁非赛马和伊犁赛马)肠道中ARG流行率进行成对比较。虽然伊犁马的样本量最大,但伊犁马中ARG的数量和丰度并不是最大的。然而,纯种马的ARG丰度和数量最高,是藏马的两倍多(图4D,E)。藏马肠道中ARG的丰度和数量最低,这可能是由于藏马在青藏高原自由放牧,很少接触抗生素。此外,虽然伊犁赛马、伊犁马、设特兰小马的遗传背景、训练模式、繁殖地不同,但这些马肠道中ARG的丰度和数量是相似的。在这六个品种中,纯种马在整个生命周期中都要接受商业配方饲料、抗生素给药、马厩和禁闭条件,这可能是纯种马群体中ARG抗生素丰度高的原因之一。
对赛马肠道中潜在提升运动能力的微生物提出新见解
Novel insights into potential performance-enhancing microbes in racehorses
为了揭示赛马肠道菌群与运动表现之间的联系,我们检查了精英赛马(n = 21,伊犁赛马)和一组年龄匹配的非赛马(n = 35,伊犁马)的微生物组之间的差异。主坐标分析(PCoA)显示,赛马和非赛马的微生物组成明显分离(图5A)。在物种水平上对两组肠道微生物组组成的差异进行了进一步分析。来自Prevotell的MAG(MAG36.bin.6,MAG266.bin.12,MAG146.bin.68,MAG122.bin.6, MAG120.bin.60),Lachnospiraceae(MAG252.bin.16,MAG32.bin.44),Phascolarctobacterium(MAG20.bin.3,MAG19.bin.10,MAG23.bin.66,MAG3.bin.34,MAG58.bin.49,MAG23.bin.2,MAG35.bin.4,MAG80.bin.33,MAG240.bin.26,MAG77.bin.65,MAG104.bin.14,MAG148.bin.27),Oscillospiraceae(MAG59.bin.38),Eubacterium(MAG77.bin.21)和Ruminococcus(MAG73.bin.20,MAG28.bin.65)在精英赛马的肠道中显示出比非赛马更高的丰度(图5B)。Lachnospiraceae和Ruminococcus是赛马中高丰度的微生物,这与Plancade等人对耐力马的研究是一致的。有趣的是,被定义为运动性能相关细菌的Lachnospiraceae,Oscillospiraceae和Ruminococcus被发现在人类运动员的微生物组中富集,这表明运动性能相关的细菌可能至少部分在动物和人类中保守。
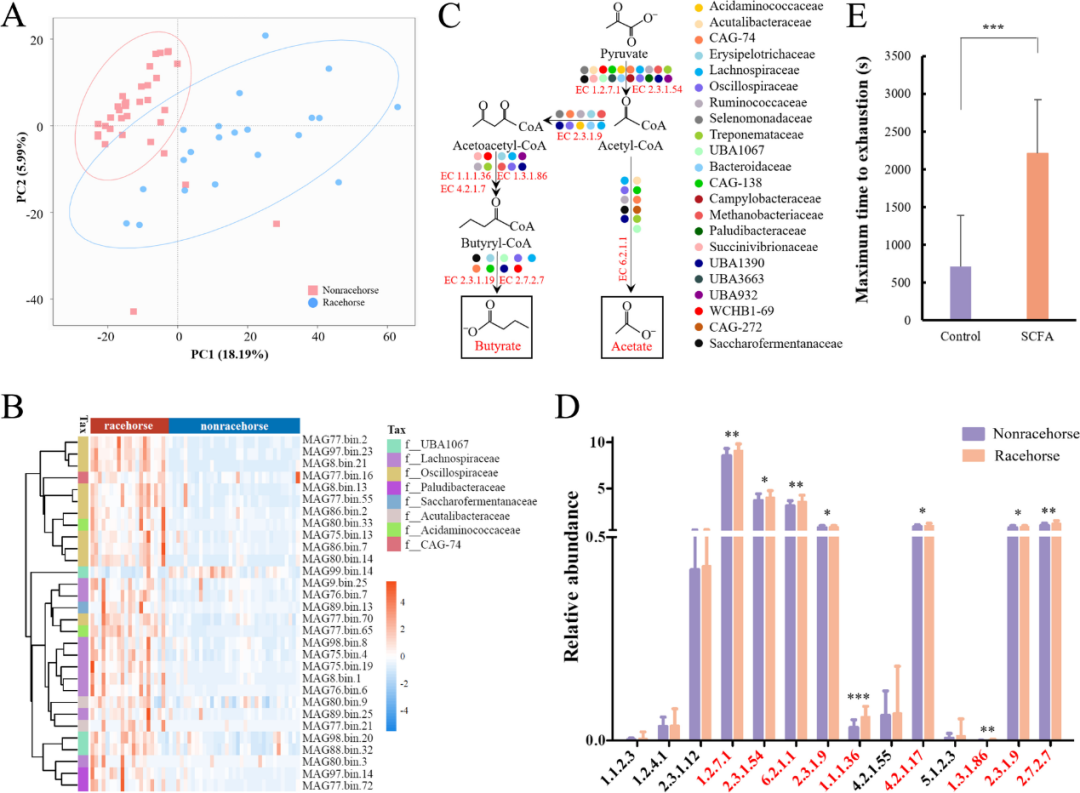
图5 赛马肠道中具有潜在提升运动能力的微生物分析。
(A) 通过PCoA可视化赛马和非赛马肠道微生物群的差异。(B) 赛马和非赛马中差异丰度最高的30个MAG。(C) 在赛马中富集的细菌在乙酸盐和丁酸盐合成途径的每一步都含有关键酶。(D) 柱形图显示了赛马和非赛马之间参与乙酸盐和丁酸盐合成的关键酶的差异。(E) 乙酸盐和丁酸盐处理显著改善小鼠的运动性能。
为了进一步探索差异丰度细菌的功能,我们比较了赛马组和非赛马组之间微生物组的代谢途径。如图5C所示,Lachnospiraceae,Phascolarctobacterium,Oscillospiraceae,Eubacterium,Ruminococcus,Campylobacteraceae, Methanobacteriaceae, Succinivibrionaceae, Bacteroidaceae, Erysipelotrichaceae和Treponemataceae都在赛马中富集(图5B),含有许多乙酸盐和丁酸盐合成途径中的关键酶。与非赛马相比,乙酸盐和丁酸盐合成途径中的关键酶(EC 1.2.7.1、EC 2.3.1.54、EC 6.2.1.1、EC2.3.1.9、EC 1.1.1.36、EC 4.2.1.17、EC 1.3.1.86、EC 2.3.1.9和EC 2.7.2.7)在赛马肠道中显著富集(图5D)。更重要的是,36个MAG(例如,属于Lachnospiraceae的MAG15.bin.4,属于Oscillospiraceae的MAG122.bin.15,属于Ruminococcus的MAG28.bin.65和属于Treponema的MAG17.bin.19)在赛马中高于非赛马。赛马中Treponem的富集与先前Plancade等人在耐力马和Janabi等人在标准竞赛马中进行的研究一致,这些研究表明训练会导致Treponem的增加。这些伊犁赛马中富集的MAG含有生产乙酸盐和丁酸盐的主要途径中的所有基因。这些结果表明,这些在赛马中富集的微生物可能在肠道中合成更多的醋酸盐和丁酸盐。然而,这里观察到的差异可能是由于赛马和非赛马的饮食、环境或运动不同。需要进一步的研究来证实这一点。
醋酸盐和丁酸盐已被证明可以调节骨骼肌功能和运动能力。一些团队报道,醋酸盐单独或与丁酸盐一起连续补充可改善耐力表现和肌肉力量。我们还证实,将SCFA(乙酸盐和丁酸盐的混合物)处理小鼠导致其最大力竭时间显著增加(图5E)。与生理盐水对照组(n = 8)相比,口服SCFA处理的小鼠平均跑步时间增加约87.23%。虽然有报道口服醋酸可以作为马休息时骨骼肌的重要能量来源,但SCFA是否能增强马的运动表现需要在未来深入研究。
- 结论 -
我们提出了马肠道微生物组的大规模宏基因组测序数据集和参考基因组组装,这将提高对未来马的微生物组研究进行分类分组以及宏基因组和宏转录组分析的能力。在MAG中鉴定出高丰度和多样性的ARG,这表明马肠道是抗生素抗性基因的储存库。此外,通过使用组装的基因组来挖掘马肠道微生物组的功能,观察到36个MAG(例如,属于Lachnospiraceae,Oscillospiraceae和Ruminococcus)在赛马中富集。这些富集在赛马中的细菌可能会产生更多的乙酸盐和丁酸盐,从而有可能提供更多的短链脂肪酸来促进运动表现。这些产生醋酸盐和丁酸盐的微生物有望被用作识别或选择耐力赛马的生物标志物,并可能被开发成益生菌,用于促进马匹的运动和健康。我们的研究为马肠道微生物群提供了详尽的参考基因组数据集,我们的结果强调了宿主和肠道微生物群之间的复杂相互作用。
参考文献
Li, C., Li, X., Guo, R. et al. Expanded catalogue of metagenome-assembled genomes reveals resistome characteristics and athletic performance-associated microbes in horse. Microbiome 11, 7 (2023). doi: https://doi.org/10.1186/s40168-022-01448-z
- 作者简介 -
第一作者

石河子大学
李村院
博士研究生
第一作者:李村院,石河子大学博士研究生,主要关注肠道微生物群落与功能,肠道宏基因组,功能基因挖掘等。目前在Science、Microbiome、Nucleic Acids Research、Genomics、Frontiers in Genetics、Research in Veterinary science、 Gene等期刊上发表多篇文章。

石河子大学
李晓悦
博士研究生
第一作者:李晓悦,石河子大学博士研究生,主要关注动物肠道微生物功能基因的挖掘。目前在Microbiome、Genomics、Functional & Integrative Genomics、Genes等期刊发表多篇文章。
通讯作者

石河子大学生命科学学院
胡圣伟
教授、副院长
通讯作者:胡圣伟,博士,石河子大学生命科学学院教授,博士生导师,生命科学学院副院长,国家级人才项目入选者,首批兵团英才第一层次入选者。目前主要从事新疆特色动物资源开发与利用研究,主持国家自然科学基金、科技部第三次新疆综合科学考察专题、农业部外来入侵物种重点调查项目等省部级以上项目11项。已在Science、Nucleic Acids Research、eLife、Microbiome等国际高水平期刊发表论文30余篇,被引用650余次。获得授权中国发明专利4项,授权美国专利1项。获得新疆生产建设兵团科技进步二等奖3项、全国大学生生命科学竞赛二等奖指导教师2次、兵团“互联网+”大学生创新创业大赛金奖指导教师、石河子大学优秀硕士论文指导教师等奖励10余项。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读

















 3336
3336

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









