






点击蓝字 关注我们
cisDynet:可用于构建基因动态调控和网络的整合分析平台

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.152
● 2023年11月19日,南京大学陈迪俊团队在iMeta在线发表了题为 “cisDynet: an integrated platform for modeling gene-regulatory dynamics and networks” 的研究文章。
● cisDynet 为研究人员提供了一个用户友好型解决方案,最大限度地减少了编程的需要,确保了结果的可重复性,并大大简化了表观基因组数据的探索。
● 第一作者:祝涛、周欣恺、由雨欣
● 通讯作者:陈迪俊(dijunchen@nju.edu.cn)
● 合作作者:王琳、赫兆辉
● 主要单位:南京大学医药生物技术全国重点实验室
亮 点
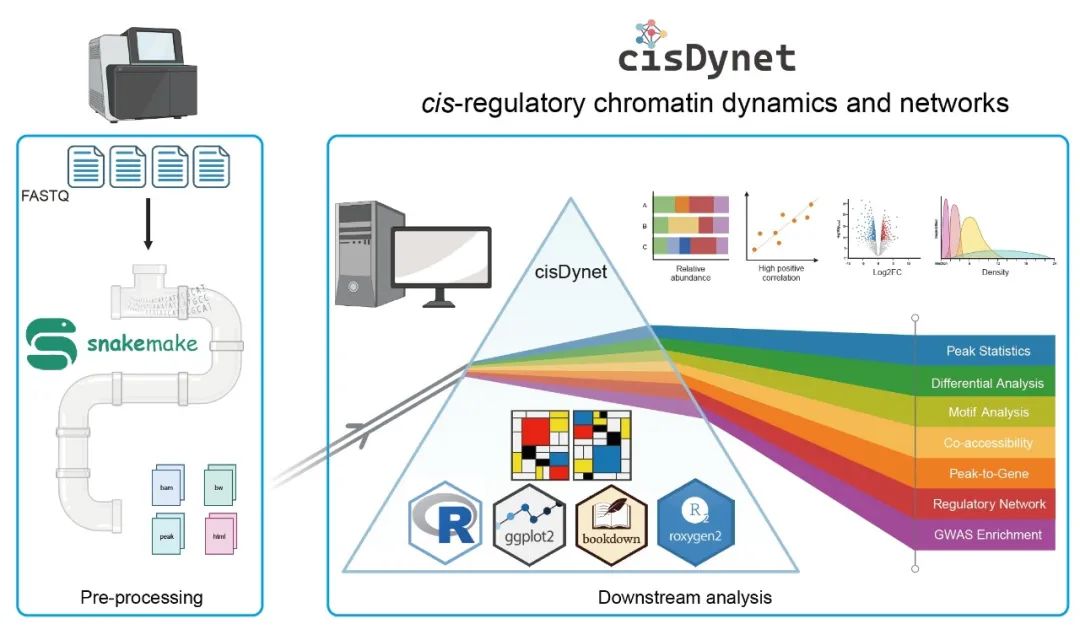
● cisDynet 可全面、高效地处理染色质可及性数据,包括预处理、高级下游数据分析和可视化;
● cisDynet 提供了一系列分析功能,如处理时间序列数据、共开放分析、将 OCR 关联到靶基因、构建顺式调控网络和 GWAS 变异富集分析;
● cisDynet 可简化组织/细胞类型特异性 OCR 或随时间发生动态变化的OCR鉴定,并有助于整合 RNA-seq 数据以描述时间轨迹。
摘 要
染色质可及性测序已被广泛用于揭示遗传调控机制和推断基因调控网络。然而,有效整合大规模染色质可及性数据集一直是一项重大挑战。这是由于缺乏全面的端到端解决方案,因为许多现有工具主要强调数据预处理,而忽略了下游分析。为了弥补这一不足,我们推出了cisDynet,这是一种整体解决方案,它使用 Snakemake 和 R 函数将数据预处理和高级下游分析功能结合在一起。此外,它还能进行复杂的数据探索,如组织特异性开放染色质区域(open chromatin regions, OCR)识别、时间序列数据拟合、整合RNA-seq数据以建立OCR与基因的关联、构建调控网络,以及对全基因组关联研究 (GWAS) 变异进行富集分析。为了验证其可行性,我们应用cisDynet重新分析了ENCODE项目中人类不同组织的 ATAC-seq数据集。该分析成功地划分了组织特异性OCR,建立了OCR与靶基因之间的联系,并将这些发现与1861个GWAS关联的变异有效地联系起来。此外,cisDynet 还有助于剖析小鼠胚胎发育的时间序列开放染色质数据,揭示了开放染色质随时间的动态变化,并确定了决定分化轨迹的关键转录因子。总之,cisDynet 为研究人员提供了一个用户友好型解决方案,最大限度地减少了编程的需要,确保了结果的可重复性,并大大简化了表观基因组数据的探索。
视频解读
Bilibili:https://www.bilibili.com/video/BV1Da4y1Z7Ph/
Youtube:https://youtu.be/edqNTge35Vg
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
在真核生物的细胞核中,DNA缠绕在核小体上形成致密的染色质结构。缺乏核小体的基因组区域被称为染色质可及区或染色质开放区(OCR)。这些染色质开放区是转录因子(TF)和 RNA 聚合酶的结合位点,在调控下游基因表达方面起着至关重要的作用。目前有多种高通量基因组学技术可识别基因组中的OCR,包括 ATAC-seq、MNase-seq、FAIRE-seq和 DNase-seq等。其中,ATAC-seq 因其实验周期短、所需起始材料少、灵敏度高等优势而被得到广泛应用。
ATAC-seq和其他染色质可及性数据的标准分析流程通常都包括开放区识别和注释、差异开放区鉴定、TF足迹分析和构建调控网络等步骤。然而,实现上述功能,通常研究人员需要使用多个软件包,而每个软件包都有其特定的输入要求,这可能会带来一些不便和挑战。到目前为止,适应各种分析场景的染色质可及性数据的综合工具包还存在明显的空白。
靶基因的确定是染色质可及性数据分析的关键步骤。在数据样本量有限的情况下,基因注释通常是将开放区与最近的转录起始位点(TSS)联系起来。一旦确定了差异开放区的目标基因,就可以将它们与从 RNA-seq 数据中获得的差异表达基因进行比较,从而进行联合分析。包括 DiffBind、HOMER等在内的一些软件包已经支持这类分析。然而,在分析多个样本的情况下,可以通过考虑不同样本中染色质可及性和基因表达的相关性来确定目标基因。这种方法已被证明有效可行。但值得注意的是,目前还没有用于进行这类分析的专用工具。
在此,我们开发了cisDynet,旨在利用染色质可及性数据为顺式调控动态和基因调控网络建模。该工具包由两个基本部分组成:基于Snakemake的数据预处理流程和提供多种功能和可视化工具的R包。除了开放区统计评估、差异开放区鉴定、motif和TF足迹活性评估等标准分析外,我们还加入了为有效处理多个样本而定制的高级功能。这些功能包括时间序列数据拟合、共开放分析、开放区与靶基因的关联、构建调控网络、GWAS 变异富集分析等。重要的是,这些功能对计算资源要求较低,而且与个人电脑兼容。为方便用户理解和使用这些功能,我们提供了一份全面的使用手册,可在以下网址查阅(https://tzhu-bio.github.io/cisDynet_bookdown/book/index.html)。此外,我们还应用cisDynet重新分析了从ENCODE项目中下载的17种人体组织的ATAC-seq数据。我们的分析成功鉴定了组织特异性开放区,建立了开放区与靶基因之间的关联,并将这些发现与 1861 个GWAS 变异有效地联系起来。最后,我们还利用 cisDynet 分析了小鼠胚胎发育的时间序列数据,揭示了开放区随时间的动态变化,并确定了决定分化轨迹的关键转录因子。总之,cisDynet 简化了染色质可及性数据分析过程,减少了大量代码开发的需要,并确保了结果的可重复性。
结果和讨论
cisDynet概览
cisDynet 包括两个核心组件:基于 Snakemake实现的数据预处理工作流程,以及为后续下游分析设计的配套R软件包。我们的数据预处理工作流程遵循 ENCODE项目推荐的标准参考流程。这一综合流程包括测序接头去除、数据比对、数据过滤、质量控制指标计算和开放区鉴定等任务。我们的数据预处理流程还支持处理基于UMI的测序策略数据。运行完预处理流程后,cisDynet会自动生成一份HTML报告。该报告为用户提供了清晰的数据质量评估结果概览,包括片段插入分布、TSS富集分数和FRiP等重要指标。
cisDynet R软件包提供了一整套用户友好型功能,涵盖了从质量控制到高级下游分析和可视化等广泛任务(图 1)。整体来看,cisDynet软件包能让用户轻松地进行各种分析,包括 (1) 评估不同开放区之间的相似性;(2) 对开放区进行注释;(3) 进行两组/三组差异开放区分析;(4) 对多个样本进行差异分析;(5) 拟合时间序列数据并整合 RNA-seq 数据,以识别动态变化的OCR和相关基因;(6) 通过足迹分析量化TF活性;(7) 对开放区进行共开放分析;(8) 探索开放区与目标基因之间关联;(9) 构建TF-TF 调控网络;(10) 遗传变异的富集分析。此外,我们的软件包还提供了超级增强子识别和峰值富集分析等任务的功能。总之,cisDynet 提供了丰富的功能,为各种分析提供了便利,这使它对专注于通过开放染色质区域揭示基因表达调控的复杂性的研究人员特别有价值。
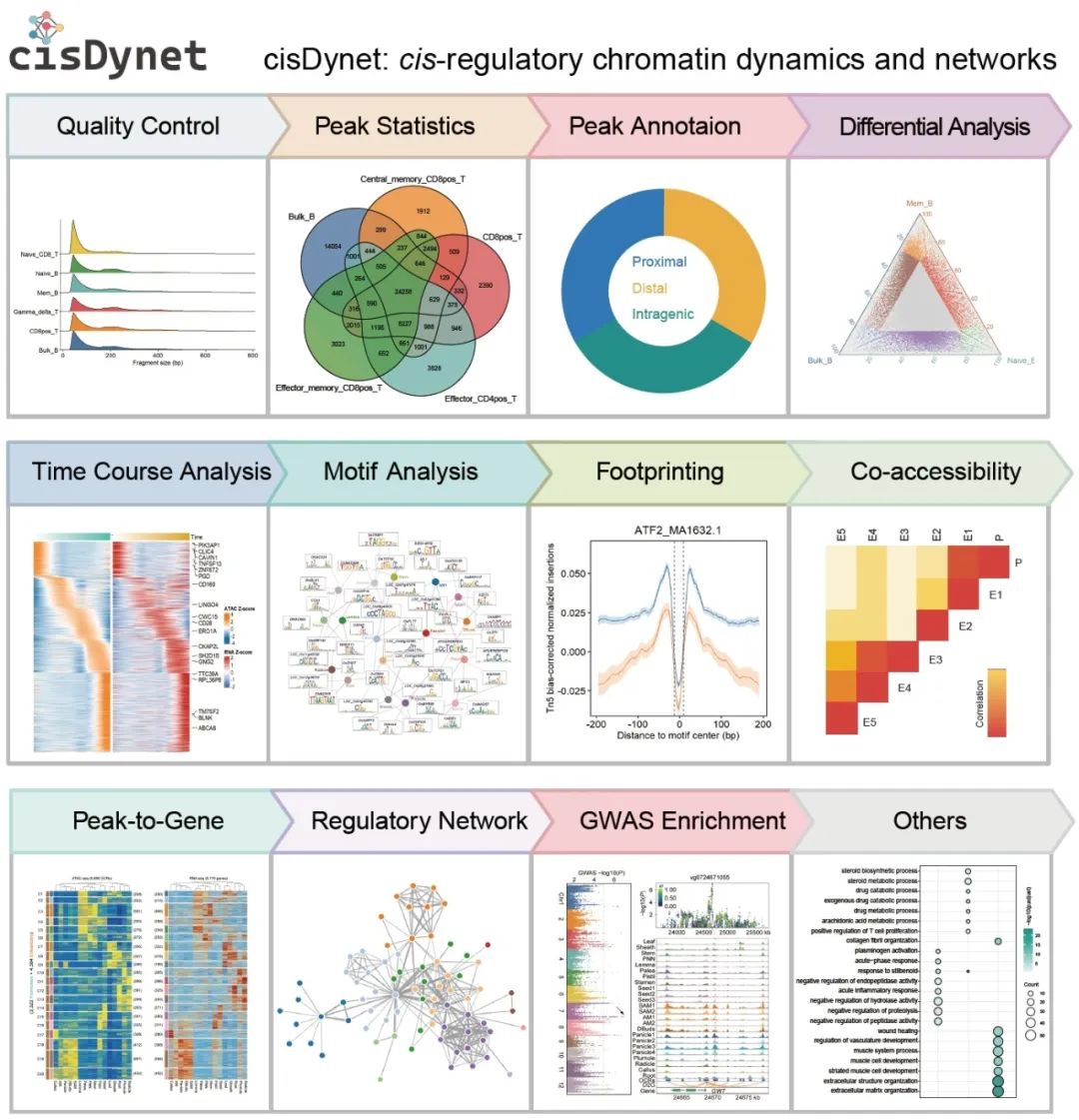
图1. cisDynet R软件包提供的染色质可及性数据分析功能模块概览
cisDynet 与其他工具的比较
目前已有几款可用于ATAC-seq数据处理的工具,包括 esATAC、ATACseqQC、AIAP 和 PEPATAC等。这些工具主要用于ATAC-seq数据的预处理,包括去除测序接头、数据比对和开放区鉴定等任务。然而,它们的主要功能仍局限于这些基础数据处理阶段(图 2)。它们对于下游高级分析的能力往往有限,例如对多个样本进行差异分析、将调控区与基因表达联系起来、构建调控网络、探索 GWAS 变异的富集等(图2A,B)。
我们开发的cisDynet提供了一个高效的分析管道,涵盖了ATAC-seq数据分析的整个流程,从基本的数据预处理到全面的下游高级分析(图2A)。cisDynet 的一个重要特点是它能整合ATAC-seq和RNA-seq数据进行联合分析,而现有软件很少具备这种功能。一般地,大多数ATAC-seq工具都遵循独立的流程:首先分别识别差异开放区和差异基因,然后尝试将开放区分配到最近的基因,再与差异基因进行交叉。然而,在处理大样本量或群体规模的数据时,这种方法就变得不那么有效了。在这种情况下,我们的cisDynet具有独特的优势。它不仅能识别差异开放区和基因,还能整合ATAC-seq和RNA-seq数据。此外,尽管TOBIAS目前是基于 ATAC-seq 数据构建调控网络的特色工具,但它侧重于利用“足迹”构建TF-TF调控网络,而没有整合RNA-seq数据。相比之下,cisDynet 在此基础上整合了RNA-seq数据,从而提高了调控网络构建的可信度。
基因组中存在一些可能产生极高信号的异常区域。这些区域大多是由于存在重复序列、基因组序列组装错误等原因造成的。在哺乳动物中,这些“问题”区域已经被推断出来并经过人工检查,并被称为黑名单,且广泛应用于基因组学数据分析。然而,目前还没有系统的植物基因组黑名单。为了填补这一空白,我们使用greenscreen软件,结合从 ChIP-Hub 数据库收集的数据,获得了五个植物物种的“问题”列表:拟南芥、水稻、玉米、大豆和番茄。我们相信,在使用 cisDynet 分析植物表观基因组数据时排除这些黑名单区域将提高定量的准确性。
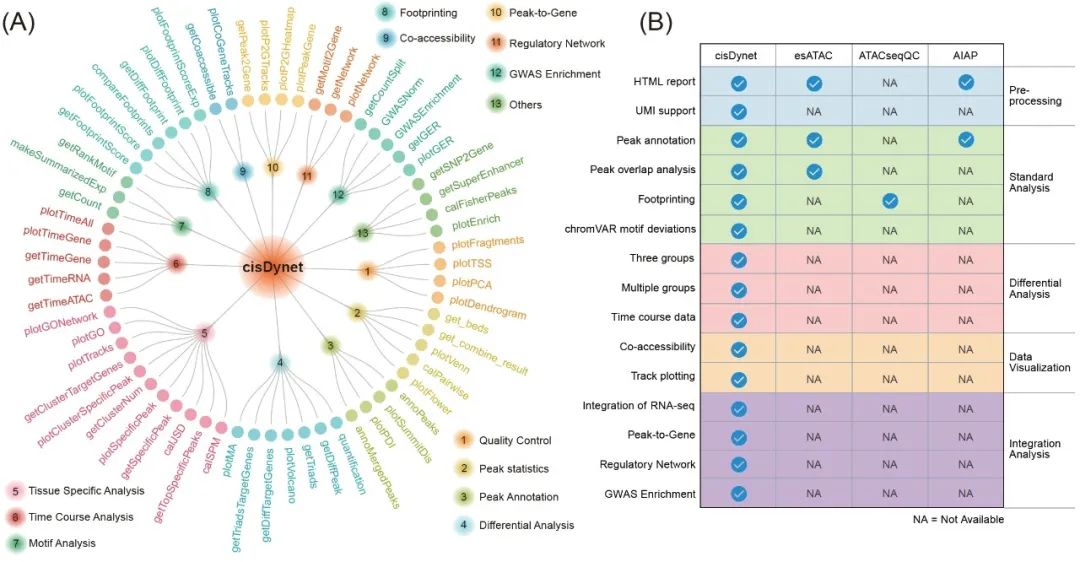
图2. cisDynet 与其他工具的比较
(A) cisDynet 所提供的不同模块的功能。(B) cisDynet 与其他用于处理染色质可及性数据的工具的比较
cisDynet 可对多个样本进行全面的差异分析
差异开放区的鉴定既有助于辨别特定条件或处理对染色质状态的影响,也有助于鉴定与这些差异开放区相关的潜在调控因子。除了cisDynet提供的广泛使用的两组别差异开放区鉴定外,我们的软件包还提供了三组和多组差异开放区分析及可视化功能(图3)。为了鉴定三组别的差异峰,我们从之前的一项研究中汲取灵感,设计了一种三元组分析策略。最初,我们构建了一个由七种类型组成的矩阵:A显性、B显性、C显性、A抑制、B抑制、C抑制和平衡(图3A)。随后,对于定量归一化的矩阵,我们计算了每个峰的定量结果与该矩阵中七个分组之间的欧氏距离。然后,我们选择欧氏距离最近的分组对该特定峰进行分类。为了直观地显示分组结果,我们使用了三元图进行可视化。我们从 ENCODE获得了人类染色质可及性数据,重点研究了肺、肝脏和脾脏,以评估这三个器官染色质可及性的差异(图3B)。值得注意的是,我们得出的不同分组与观察到的定量区别完全一致,从而验证了我们分类的准确性(图3C)。
对于三个以上组别之间的差异分析,可从归一化矩阵中对每个开放区计算香农熵指数。该熵指数可用于筛选和识别不同组间高度可变的峰值。值得注意的是,这种筛选方法可能会导致样本间差异峰的数量不平衡,尤其是当大多数样本显示出相对较高的相似性,而少数样本与其他样本存在显著差异时。为了说明这一现象,我们利用了多个免疫细胞系的ATAC-seq 数据。与其他细胞系相比,我们在浆细胞树突状细胞(pDCs)和单核细胞中观察到了明显的分化。由于大多数样本由T细胞组成,我们的分析主要得到了与pDCs和单核细胞相关的差异峰,因此很难辨别不同T细胞群之间的差异。为了应对这一挑战,我们采用了一种名为特异性衡量(SPM)的额外指标。SPM 可以识别和过滤单个样本或多个样本中高度开放的区域。结果表明,这种方法大大提高了我们比较样本间可及性差异的能力。我们将这一策略应用于ENCODE数据,以确定不同器官之间的不同 OCR(图 3D)。这些器官特异性OCR主要位于TSS远端区域,这与之前的研究相似(图 3E)。cisDynet还提供了研究与这些差异OCR相关的生物通路的功能(图3D、F)。cisDynet 提供的信号文件也可以直接通过表观基因组浏览器(如WashU Epigenome Browser)进行可视化。例如,HOXA11是子宫上皮细胞和基质细胞中特异表达的基因(图3G)。HOXA13是另一个在结肠中高度特异性表达的基因,影响表皮分化(图3G)。其他例子包括SNAI2和TBX15,它们都在腓肠肌内侧表现出特异性开放(图3G)。
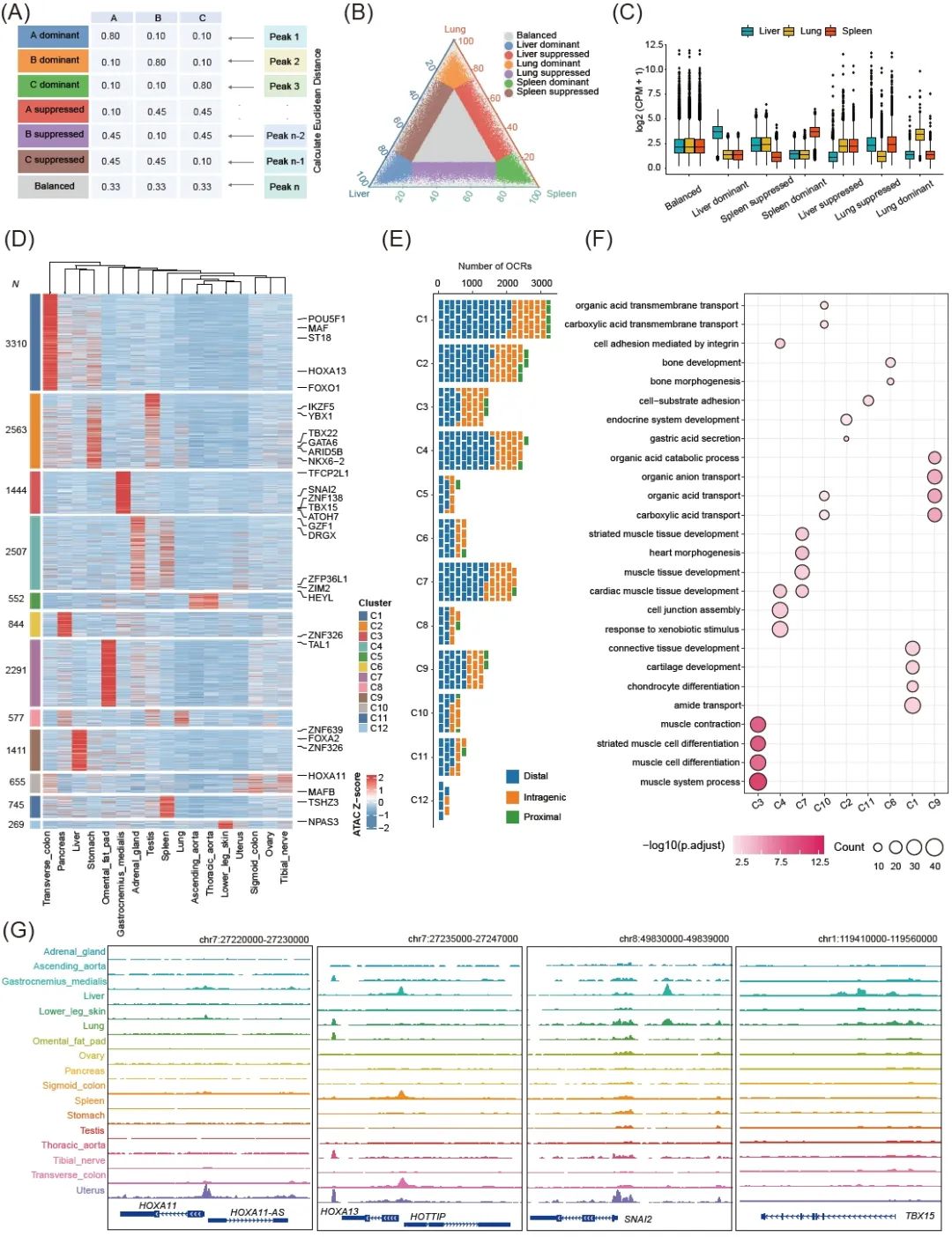
图3. 使用 cisDynet 比较染色质可及性差异并识别组织特异性开放区的示例
(A) 使用七种类型的开放区预定义矩阵对三个样本的染色质可及性差异进行分类。(B)三元图显示肺、肝和脾三个器官的染色质可及性差异。(C)箱线图展示了(B)中七个分类的染色质开放程度。(D)利用cisDynet鉴定出的组织特异性开放区。根据染色质可及性的相似性,所有组织特异性开放区被分为 12 个簇。(E) (D)中每个簇中所包含的基因内部、近端和远端开放区的比例。(F)每个簇的开放区靶基因所涉及的生物学通路的富集分析。(G)基因组浏览器展示了ATAC-seq信号在 HOXA11、HOXA13、SNAI2 和 TBX15 位点附近的分布。
用 cisDynet 描述时间轨迹数据
时间序列实验对于揭示细胞动态过程(如细胞分化或对压力的反应)至关重要。与单细胞拟时间分析不同,bulk数据不需要推断其时间顺序。我们只需要根据实验设计中定义的时间点来拟合动态变化的OCR。首先,可以使用上述差异分析方法识别出不同时间点上高度动态的OCR。然后,我们应用局部多项式回归拟合来建立时间点与这些动态OCR之间的关系。通过拟合,我们可以根据拟合模型预测OCR值随时间的平滑变化(图 4A)。作为概念验证,我们利用小鼠胚胎干细胞(mESCs)分化过程中五个时间点收集的ATAC-seq数据进行了分析(图4B)。我们总共获得了2165个随时间动态变化的OCR,并整合RNA-seq数据找到了这些OCR的相应靶基因(图4C)。通过这一分析,我们观察到Sox2和Ackr3等基因参与其中,众所周知,这些基因在维持神经干细胞和祖细胞的活性方面发挥作用(图4D)。相反,Sox11和Gfap等基因分别在中期和晚期发挥重要作用,其中Gfap是星形胶质细胞的标记基因。
mESCs 在两天后会分化成三个亚群,这促使我们研究驱动这一过程的关键调控因子。我们首先利用TOBIAS衡量了这五个阶段中所有TF的全基因组活性,并将其称为足迹分数。然后,我们计算了足迹分数与基因表达之间的皮尔逊相关系数,并根据相关系数确定了驱动调控因子(图4E)。我们比较了五个时间点的TF活性,发现 JUNB 和 Mlxip 在第2天和第4天的活性较高。正如我们所预期的那样,还观察到JUNB和Mlxip在第 2 天和第 4 天分别有更深的足迹(图4F)。此外,我们还观察到POU3F1在早期阶段具有很强的活性,而该基因被报道与神经发育有关。相反,NFIB 在第20天有明显的高活性,这在最初的研究中也被确定为驱动调控因子。
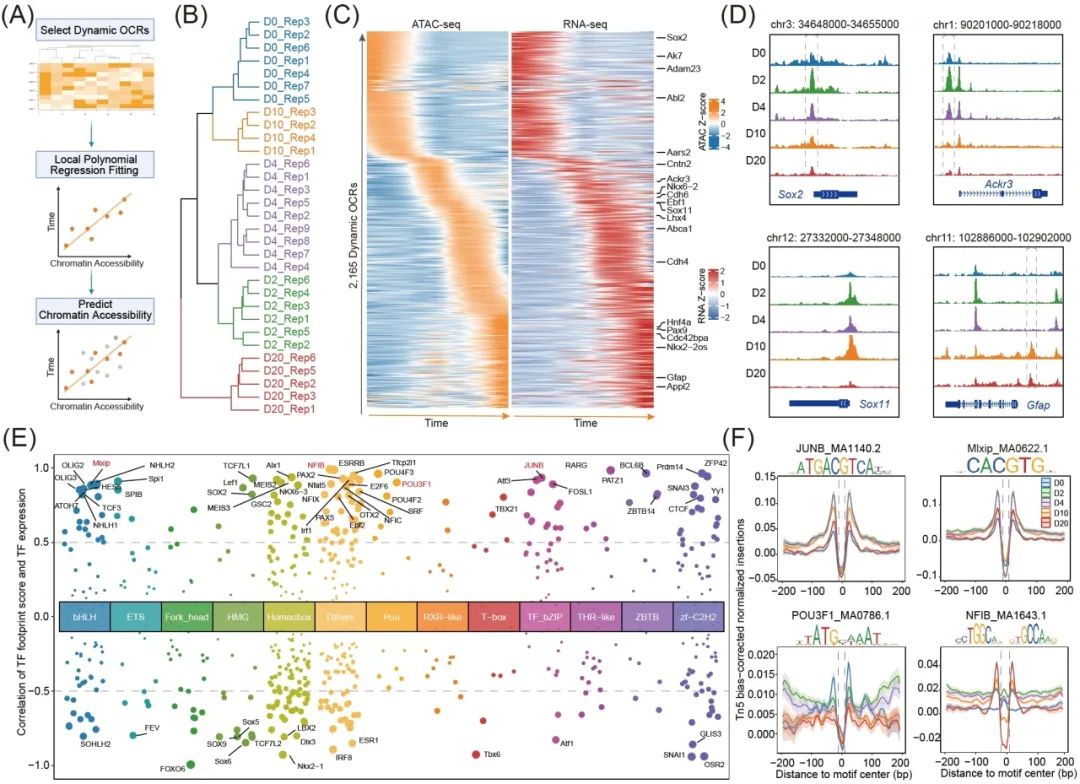
图4. 通过cisDynet整合RNA-seq鉴定随时间动态变化的染色质开放区和参与小鼠胚胎干细胞(mESCs)发育过程的重要转录因子。
(A) cisDynet识别随时间动态变化的开放区的流程图。(B) 不同时间点染色质可及性数据的聚类结果。(C)使用cisDynet鉴定出的 2165 个随时间动态变化的开放区及其靶基因的表达变化。目标基因的确定原则是它们与开放区最近,并且与其表达的皮尔逊相关系数不小于 0.7。(D) 基因组浏览器轨迹图显示了 mESCs 分化过程中 Sox2、Ackr3、Sox11 和 Gfap 基因附近开放区的动态变化。(E)散点图展示了不同基因家族TFs的足迹得分与其自身表达之间的皮尔逊相关系数分布。少于 15 个 TFs 的家族被合并到其他家族中。TF足迹分数由TOBIAS计算得出。(F) 在 mESCs 分化过程中的四个时间点,Tn5切点在TF足迹周围的分布。
cisDynet可有效识别影响基因表达的调控区域
大量研究表明染色质可及性直接影响基因的表达水平,并且染色质可及性与基因表达之间存在很强的正相关性。这促使我们在全基因组范围内预测OCR的潜在靶基因。由于染色质的空间构象,OCR可能不会线性调节最近的基因。根据三维相互作用的信息,我们可以限制与TSS的最大调控距离(例如,人类的上下游距离为500 kb)(图5A)。我们的cisDynet可以有效地帮助每个基因在指定范围内找到潜在的OCR。以 ENCODE 数据为例,我们将潜在调控区域限定在500 kb以内。我们共发现了236,792个可靠的调控关系,其皮尔逊相关系数绝对值 (|R|) > 0.4,FDR < 0.05(图5B)。正如我们所预期的那样,相关系数随着OCR与TSS之间距离的增加而降低。此外,我们的研究结果表明,开放区到基因的关联更常见于近端OCR。每个基因平均与7.96个OCR相关联,而每个OCR平均与2.78个基因相关联。有趣的是,我们注意到与基因相比,TFs 拥有更多的开放区到基因的关联,这意味着TFs可能有着更复杂的调控机制。同时,对于那些受多个调控开放区调控的基因或TFs,它们主要富集于“分化”和“发育”生物通路。
为了进一步验证开放区-基因连接的可靠性,我们将获得的开放区-基因连接与来自不同人体组织的eQTL数据进行共定位分析。我们发现这些已确定的开放区到基因连接明显富集了eQTL,而且可以观察到组织特异性富集模式(图 5C)。例如,位于IRF8下游约80 kb处存在一个eQTL(rs16940186)。先前的研究表明,该eQTL主要在胃上皮细胞类型中发挥重要作用。我们发现了IRF8与该 eQTL 所在的OCR正好也被我们的开放区-基因连接所包含(R = 0.61,FDR = 3.8e-03),并且该OCR在胃中特异开放(图 5D)。
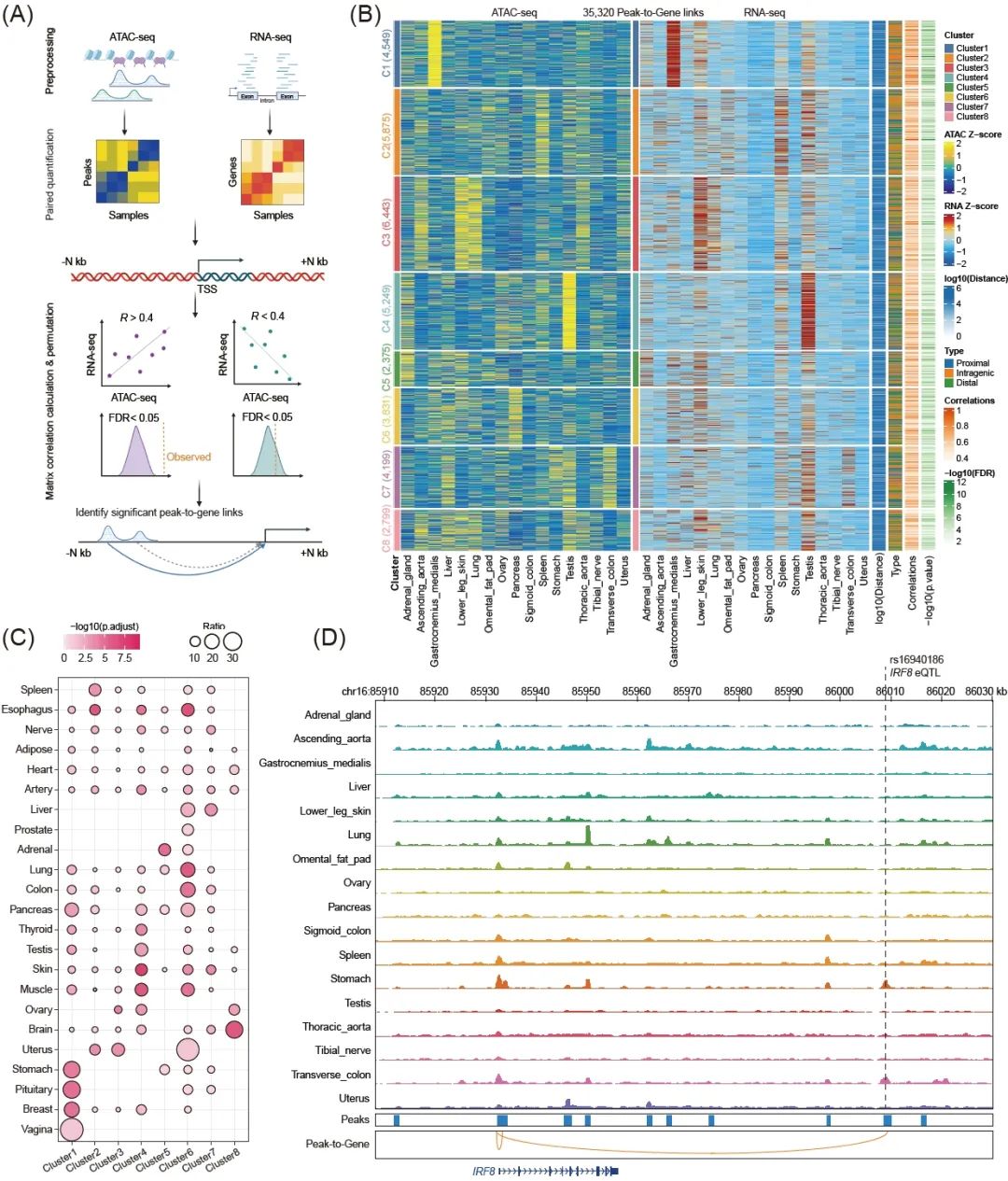
图5. 将开放染色质区域分配给目标基因
(A) 建立开放区与目标基因之间相关性的计算方法流程图。(B)热图展示了cisDynet计算出的 35,230 个开放区到基因的链接(R > 0.4,FDR < 0.05)。仅显示每个基因上下游 50 kb 范围内的峰对基因链接。每行代表一个开放区或基因。根据染色质可及性的相似性,将所有开放区分成8个簇。(C)气泡图展示了(B)中每个簇的峰到基因链接与已发表的eQTL数据的富集程度。富集程度通过超几何检验计算。(D)基因组浏览器截图显示,在 IRF8 下游约 80 kb 处存在一个 eQTL(rs16940186),它恰好位于峰到基因所在的开放区中。
全基因组关联研究(GWAS)有助于确定与各种表型和疾病相关的位点或基因。然而,这些相关位点往往位于非编码区。目前,已有几种工具可通过评估在GWAS中发现的因果变异是否富集在调控元件中,从而将组织/细胞类型与疾病和性状联系起来。然而,这些富集工具主要依赖于开放区与变异之间是否重叠,缺乏对开放区定量信息的考虑,在捕捉样本间的微小差异方面表现出有限的灵敏度。为解决这一问题,Soskic等人开发了一种名为CHEERS的工具,该工具根据开放区的定量信息将性状与细胞类型联系起来。这种方法与早先的研究结果一致,即与GWAS相关的变异往往位于组织/细胞类型特异性调控区域。根据这一原则,我们在cisDynet中引入了组织特异性得分,以适应样本间存在微小差异的分析情景(图6A)。为了验证该方法的可行性,我们从NHGRI-EBI GWAS数据库中获取数据,并筛选出显著相关变异位点数不少于 20个的GWAS,最终得到2498个GWAS。我们利用不同组织的ENCODE染色质可及性数据建立了组织与不同GWAS变异的关联。纵观这些富集结果,我们发现有1861个(74.5%)GWAS至少在一个组织中被显著富集(p < 0.05),而且确实呈现出组织特异性模式(图6B)。在没有任何先验知识的情况下,这种关联在一定程度上符合当前的生物学认知。例如,COVID-19与肺相关,脂肪肝与肝相关,卵巢癌与卵巢相关(图 6C)。这些结果证明了cisDynet在组织/细胞类型与GWAS变异之间建立关联的能力。
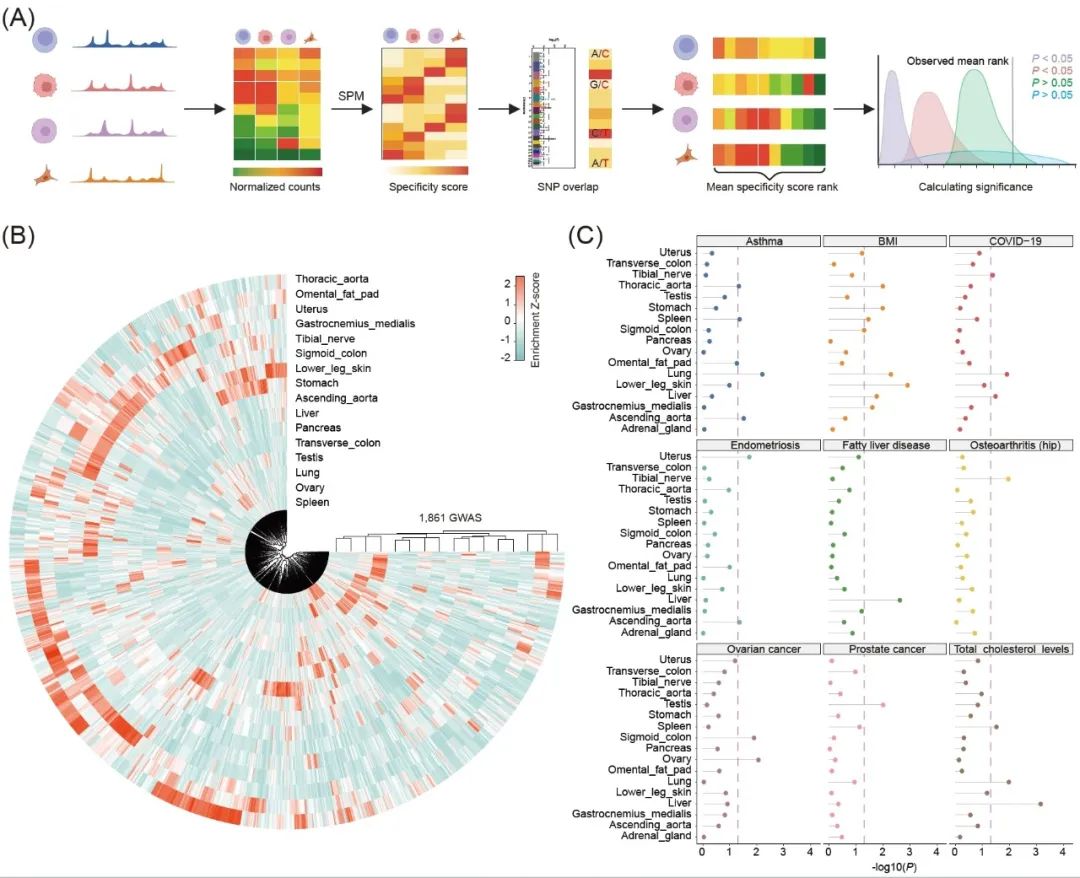
图6. 利用cisDynet推断与复杂疾病相关的组织类型
(A) cisDynet富集全基因组关联研究变异的原理示意图。(B) 热图展示了全基因组关联研究中报告的 1861 个复杂性状至少在一个组织中显著富集。(C)不同组织中开放染色质区域中九个 GWAS相关变异的富集情况。虚线表示 p = 0.05。
结 论
染色质可及性测序能让我们识别基因组上的开放区域,为了解调控元件提供有价值的信息,并能构建错综复杂的调控网络。完成这些分析往往需要使用多种工具,但工具之间相互独立,而且需要兼容各种工具的运行环境,这给研究人员带来了一定的困扰。我们开发的 cisDynet 提供了一个全面的分析流程,涵盖了从数据预处理到下游高级分析的整个过程。我们提供了详细的使用指南,其中涵盖了处理染色质可及性数据的大多数使用场景。值得注意的是,我们的cisDynet包含目前其他工具包所不具备的功能,如组织/细胞类型特异性分析、时间序列数据拟合、共开放分析以及整合RNA-seq数据以关联OCR和基因。除了分析染色质可及性数据外,cisDynet还可用于分析ChIP-seq、CUT & Tag等其他表观基因组数据分析。我们相信,cisDynet 将帮助研究人员加速表观基因组学的研究。
数据可用性声明
基于Snakemake 的预处理流程存储在https://github.com/tzhu-bio/cisDynet_snakemake。R 软件包 cisDynet 的代码存储在 https://github.com/tzhu-bio/cisDynet 。R 软件包 cisDynet 的详细使用说明请访问 https://tzhu-bio.github.io/cisDynet_bookdown/。有关结果的数据可通过 https://figshare.com/articles/dataset/Source_Data_For_cisDynet_/24498955访问。
引文格式:
Tao Zhu, Xinkai Zhou, Yuxin You, Lin Wang, Zhaohui He, Dijun Chen. 2023. “cisDynet: An integrated platform for modelinggene‐regulatory dynamics and networks.” iMeta e152. https://doi.org/10.1002/imt2.152
作者简介

祝涛(第一作者)
● 南京大学生命科学学院生物信息学专业博士在读。
● 致力于调控组学研究和相关工具开发。已连续两年获得博士研究生国家奖学金,并获得江苏省研究生创新计划、南京大学研究生标兵等多项荣誉。目前已在Nature Communications (2022a/2022b)、iMeta、Journal of Genetics and Genomics等杂志发表学术论文5篇。

周欣恺(第一作者)
● 南京大学生命科学学院生物信息学专业硕士在读。
● 目前研究方向为多组学大数据分析与挖掘及大数据整合,相关学术成果已发表于Nature Communications、iMeta、Nucleic Acids Research、Journal of Genetics and Genomics等期刊。

由雨欣(第一作者)
● 南京大学生命科学学院生物信息专业硕士在读。
● 研究方向为多组学数据分析与植物调控组学,相关学术成果已发表于iMeta等期刊。

陈迪俊(通讯作者)
● 南京大学副教授、博士生导师,江苏省特聘教授、南京大学登峰B人才支持计划入选者。
● 2003-2008年在哈尔滨医科大学接受本科教育并获得生物信息学学士学位,2017年在德国哈雷-维滕贝格大学获得博士学位,先后在浙江大学、波茨坦大学和洪堡大学从事研究工作,2019年底入职南京大学,担任生物信息学课题组PI,主要研究方向是功能基因组学和人工智能生物学。共发表学术论文近60篇,参与编写专著5部,其中以第一或者通讯(含共同)作者在Nat Commun (5篇)、Nat Neurosci、Nat Plants等主流期刊发表论文近30篇。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
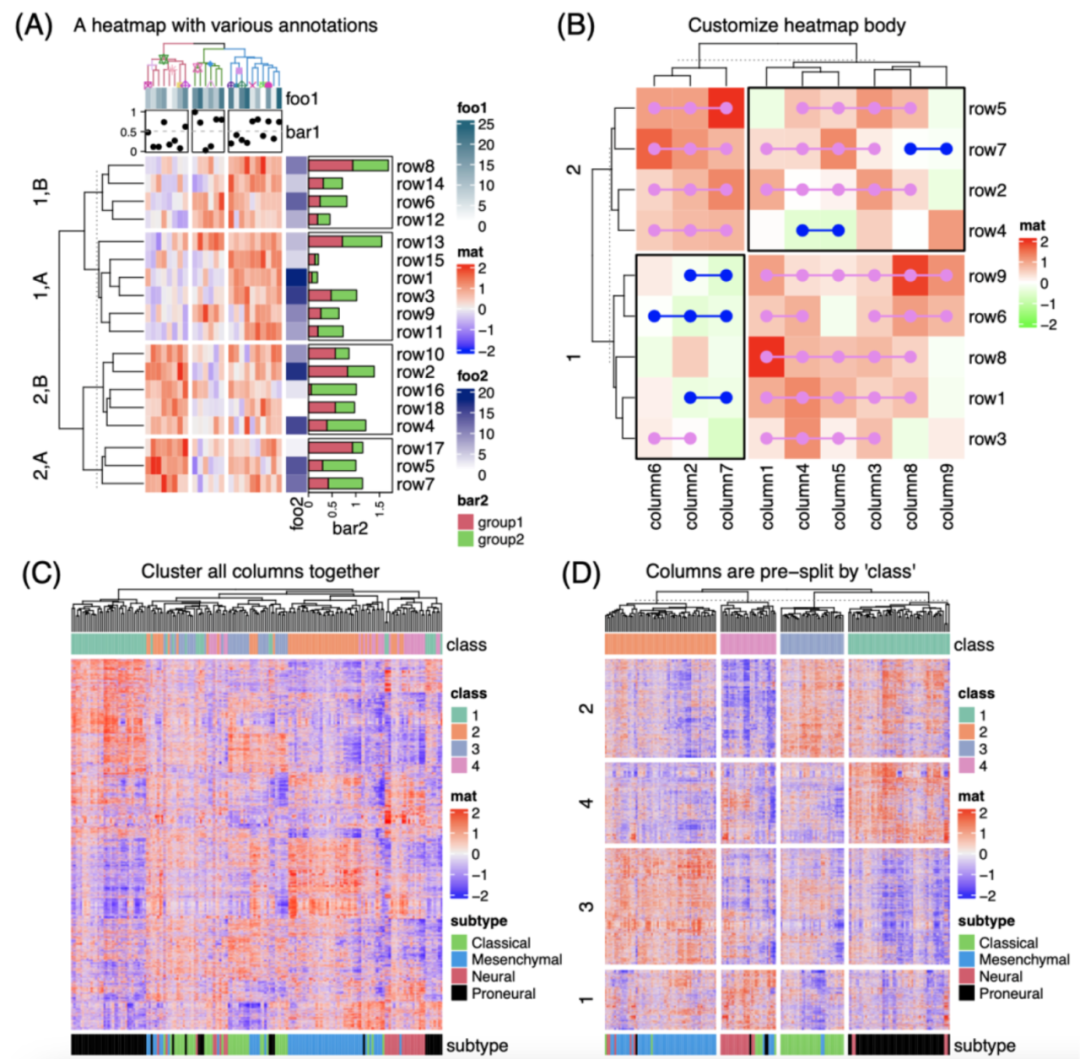
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 1260
1260

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









