






点击蓝字 关注我们
微科盟生科云(Wekemo Bioincloud):一个专为宏组学数据设计的用户友好型在线分析平台

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.175
● 2024年2月13日,中国农业科学院深圳农业基因组研究所刘永鑫团队和深圳微科盟科技集团有限公司在iMeta在线联合发表了题为 “Wekemo Bioincloud: A user-friendly platform for meta-omics data analyses” 的研究文章。
● 本文开发了微科盟生科云(Wekemo Bioincloud)——一个专业的宏组学(meta-omics)数据分析平台,该平台提供了全面的分析解决方案,方便用户在处理大量组学数据中选择适用的工具。
● 第一作者:高云云
● 通讯作者:刘永鑫(liuyongxin@caas.cn)
● 合作作者:张国兴、江舜尧
● 主要单位:中国农业科学院深圳农业基因组研究所、深圳微科盟科技集团有限公司
亮 点
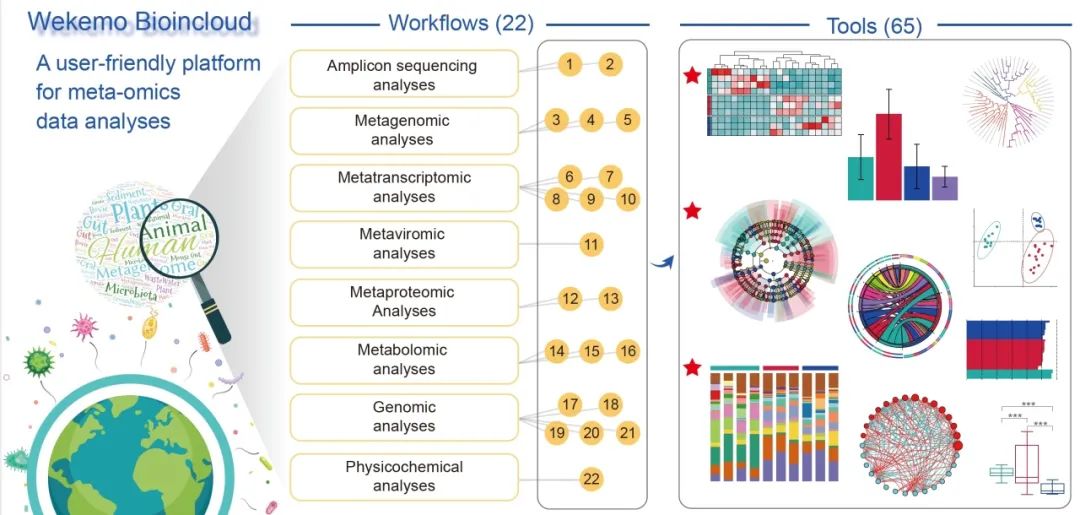
● Wekemo Bioincloud为宏组学数据提供了多种云流程和多样工具,用于数据的分析和可视化;
● 可以在线编辑矢量图,协助用户获取符合文章出版的高质量图片;
● 平台界面会展示各个工具的使用热度,提升了平台的互动性与灵活性。
摘 要
随着宏组学(meta-omics)方法在研究微生物结构、功能和互作等方面的广泛应用,我们积累了大量宝贵的数据资源。然而,如此丰富的人类/环境微生物组数据也为现有的生物信息学工具提出了新的挑战。因此,我们开发了微科盟生科云(Wekemo Bioincloud)——一个专业的宏组学(meta-omics)数据分析平台。该平台提供了全面的分析解决方案,方便用户在处理大量组学数据中选择适用的工具。目前Wekemo Bioincloud已配备了22个工作流程和65个可视化工具,可用于研究不同的数据集,是一个便于用户使用的平台。此外,该平台支持在线修改矢量图,并通过独立的注册账号,确保了用户的隐私和数据的可追溯性。Wekemo Bioincloud可以通过以下链接获取:https://www.bioincloud.tech/。目前“Wekemo”已经被引用300余次(Google Scholar,截止2024年2月),欢迎大家使用本平台并正确引用。
视频解读
Bilibili:https://www.bilibili.com/video/BV1Am411S7Uv/
Youtube:https://youtu.be/q83Igh4uCM4
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
宏组学方法的最新发展,涵盖了宏基因组学、宏转录组学、宏蛋白质组学、宏病毒组学、代谢组学以及理化数据,标志着我们正迎来全面理解复杂生物系统的革新时代。这些多维度的组学数据集包括了微生物群落的丰富性,迫切需要强大且用户友好的生物信息学工具,以揭示微生物群落的整体情况,并阐明环境与微生物组之间的相互作用。因此,微生物组领域已开发了多个数据分析平台,用于高通量组学数据的处理。例如,QIIME2和EasyAmplicon用于扩增子数据分析,Trimmomatic或fastp用于数据质控,Kraken2用于物种分类,HUMAnN3用于功能分析,MultiPrime用于高效设计引物,ImageGP用于数据可视化等等。这些工具的开发为人们深入探索组学数据提供了重要支持。
通常,标准的组学数据分析流程包括原始数据处理、物种分类注释、序列功能分析和统计分析。尽管本领域已经开发了多个分析流程/软件来标准化这些分析,然而,个性化分析和定制分析对不同的研究目的至关重要。目前,本领域已发展了各种工具、分析流程或在线分析平台来支持组学数据的分析,例如,QIIME2是一款专门为扩增子测序分析设计的软件,目前其应用也升级扩展到宏基因组数据分析;EasyAmplicon是专门用于本地服务器上扩增子测序分析的流程;MicrobiomeAnalyst是主要用于扩增子测序、宏基因组和代谢组学分析的在线平台;Notame专注于代谢组学分析;MetaProteomeAnalyzer用于宏蛋白质组数据分析;此外,还出现了许多创新方法,用于从组学数据中识别可靠和稳定的生物标志物,也有部分研究/综述对现有各种R包或软件进行总结和比较。这些工具大多数都针对一种或两种特定类型的组学数据,而当前多组学分析对于科学问题的研究至关重要。然而,多样且复杂的分析方法和工具意味着研究人员不仅需要大量时间安装各种工具或R包,而且还要适应不同工具或平台的使用,这凸显了开发适用于宏组学/多组学数据分析和可视化的在线平台的重要性。
因此,我们推出了Wekemo Bioincloud,专为宏组学数据分析量身定制,旨在解决用户在工具选择上面临的挑战。Wekemo Bioinclud平台安全地将用户的测序存储在云端,并提前配置了各种分析工具,以满足用户挖掘数据的需求。该平台包含云流程(Workflow)和工具(Tools)两个模块,并确保了用户在不同步骤具有选择性,为用户对所获数据的深入探索提供保障。可通过访问以下链接,了解平台详情https://www.bioincloud.tech/。
结 果
Wekemo Bioincloud概述
Wekemo Bioincloud包含两个主要模块:云流程模块和工具模块。在云流程模块中,用户可以逐步分析组学数据,流程会生成详细报告,说明每次分析所使用的软件和参数报告;而在工具模块,用户可以参考我们提供的演示(Demo)数据使用不同工具。该平台旨在方便研究人员访问分析,可以一键修改样品分组或一键运行所有分析。此外,平台不仅在线提供SVG编辑器,而且允许用户设置电子邮件提醒,以节省科研者的宝贵时间。平台还提供了教学视频,涵盖工具使用、云流程和结果解读,增强用户对其数据的理解。独立的注册系统确保了用户数据的隐私性、追溯性和协作性。研究人员可以通过两种方式使用平台:1) 参考在线示例(Demo)表格,选择工具模块进行数据分析;2) 提交原始数据,后台提供标准分析表,然后用户可以通过云流程或工具模块进行个性化分析。工具模块的数据每天会被删除,云流程分析的原始数据和结果报告将保留2年,为用户提供充足的时间深入挖掘数据。
截至2023年12月6日,我们对Google Scholar上引用‘Wekemo Bioincloud’的所有出版物进行了统计,删除未知/重复条目后,共计140篇文章使用了我们的平台。有趣的是,42.14%的文章使用了云流程,57.86%的文章则选择了功能模块来可视化结果。其中,相关性检验、正交偏最小二乘判别分析(OPLS-DA)、主坐标分析(PCoA)和线性判别分析效应大小(LEfSe)分析是工具模块最常用的工具。
云流程介绍
云流程模块目前已更新了22个数据分析流程,涵盖了宏基因组、宏转录组、宏蛋白质组、宏病毒组、代谢组、基因组和理化数据等各种类的型数据进行一步式分析(图1)。每个云流程都配有全面的演示报告、示例流程和结果解释,方便新用户快速入门。对于常规组学分析,用户只需参考我们的演示流程,准备原始测序数据和样本信息(metadata)。面对大量宏组学分析软件,我们的云流程还包括各种软件选项,用户可以根据自身数据的特点轻松选择不同的分析算法/软件,所有处理方法都将在输出报告中展示。

图1. Wekemo Bioincloud的22个云流程框架图
Wekemo Bioincloud平台为宏组学(meta-omics)数据分析提供了多样的云流程(Workflow),满足用户开展标准化和个性化分析。
此外,我们还为用户提供一些灵活的选择,以获得个性化的分析。例如,宏基因组流程可以对微生物组样本的肠型进行分类,宏基因组分箱流程可预测原噬菌体或分泌蛋白;10×单细胞转录组云流程整合了比对、定量、聚类和基因表达分析等过程;此外,我们还包括了纯二代组装或二、三代混合组装的方法来比较分析细菌和真菌基因组的组装效果,为系统探索数据并揭示生物学意义提供支持。
可视化工具介绍
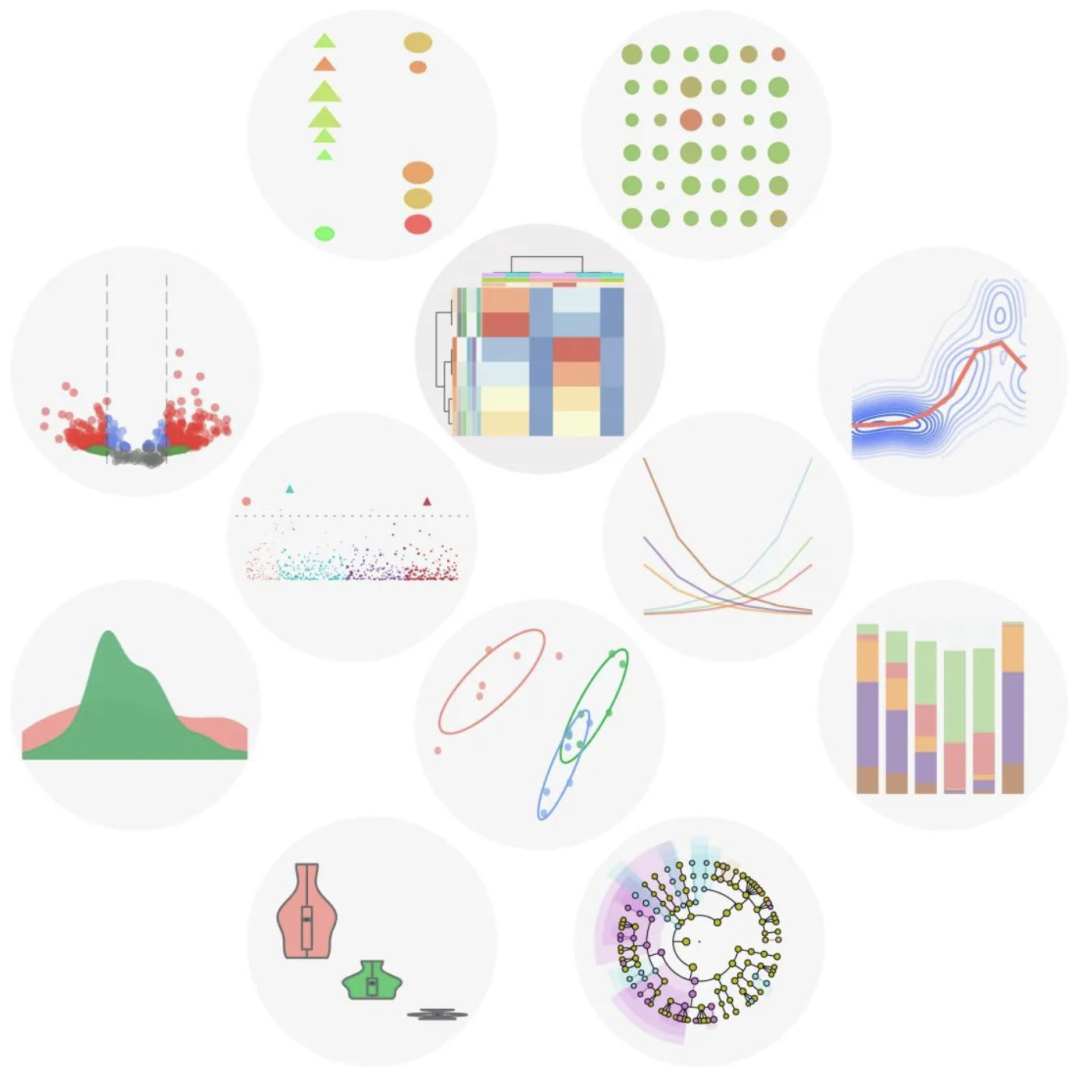
为直观呈现科学发现,Wekemo Bioincloud平台提供了一系列用于可视化、分析和比较组学数据的工具(图2)。目前已推出65个子功能,涵盖以下方面:(1) 组间特征/基因的贡献/丰富度/组成展示;(2) 组间差异比较;(3) 整体结构差异比较;(4) 数据分析,如显著性检验(ANOVA、Kruskal Wallis等);(5) 功能/代谢通路预测;(6) 基因表达差异;(7) 系统发育构建;(8) 相关性检验;(9) 可视化流程;(10) 其他(如引物设计等)。所有子功能都带有流行度和难度分数,可以帮助用户衡量每个工具的使用频率和复杂性。截至2023年12月10日,分组聚类热图、LEfSe图和分组百分比堆积条形图是目前最受欢迎的三种工具,其使用次数分别达到36,098次、34,261次和33,341次。
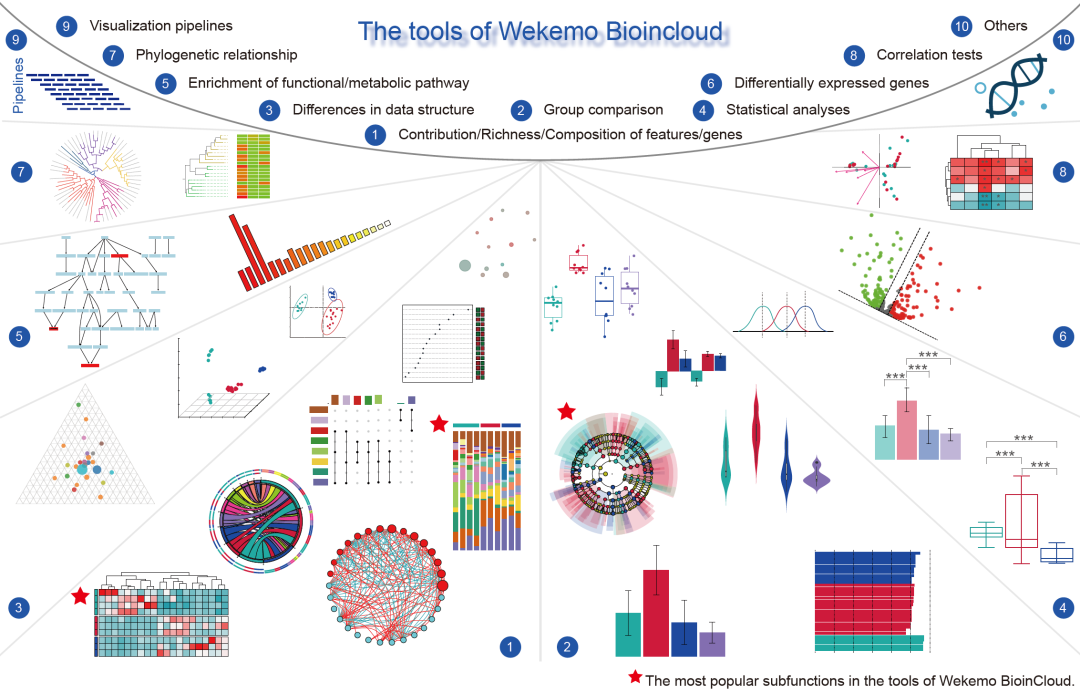
图2. Wekemo Bioincloud的工具模块可获图片的部分示意图
工具(Tools)模块根据目的可以分为10大类65种工具,10大类分别为1) 组间特征/基因的贡献/丰富度/组成展示;2) 组间差异比较;3) 整体结构差异比较;4) 数据分析,如显著性检验(ANOVA、Kruskal-Wallis等);5) 功能/代谢通路预测;6) 基因表达差异;7) 系统发育构建;8) 相关性检验;9) 可视化流程;10) 其他(如引物设计等)。
工具模块通过其65个可视化和分析功能,满足了组学研究人员的日常需求,包括散点图、条形图、气泡图、小提琴图、网络图、三元图、火山图、花瓣图、热图、路径图等(图2)。此外,还提供方差分析(ANOVA)、Kruskal-Wallis和Dunn检验等基本显著性检验,以及16S核糖体RNA(rRNA)基因比对和引物设计等常见分子生物学工具。此外,它还提供将SVG转换为各种格式(包括PDF、JPG、PNG等)的功能,Wekemo Bioincloud网站提供了工具详细解释和Demo参考数据,帮助用户快速入门。
案例1:宏基因组数据分析流程
该平台提供了三种用于分析宏基因组数据的云流程:基于参考的(reference-based)流程、从头组装(de novo)流程和分箱(binning)流程(图3)。所有原始读序(raw reads)均使用KneadData处理以获得干净读序(clean reads),使用Trimmomatic用于去除接头和低质量reads,并使用Bowtie去除宿主基因组污染。此后,过滤后的数据(clean data)可根据研究目的进行下一步处理。
目前,平台采用了宏基因组分析中12个常用的数据库来预测序列功能。
● 抗生素抗性基因数据库(Antibiotic Resistance Genes Database, ARDB):用于追踪抗生素抗性基因。
● 抗菌剂和金属抗性基因数据库(Antibacterial Biocide and Metal Resistance Genes Database, BacMet):用于明确基因对金属或抗菌剂的抗性。
● 碳水化合物活性酶(Carbohydrate-Active EnZyme, CAZy):用于描述负责裂解或构建复杂碳水化合物的酶家族。
● 综合抗生素耐药性数据库(Comprehensive Antibiotic Resistance Database, CARD):用于识别抗生素耐药性和毒力因子。
● 蛋白质直系同源簇(Clusters of Orthologous Groups of proteins, COG):尝试对蛋白质进行系统发育分类。
● 酶学委员会(Enzyme Commission, EC):编号代表酶和酶基因的进化基因谱系。
● 基因本体论(Gene Ontology, GO):注释报告基因产物与生物类型之间的联系。
● 京都基因与基因组百科全书数据库(Kyoto Encyclopedia of Genes and Genomes, KEGG):用于识别生物系统内的功能。
● 移动遗传元件(Mobile Genetic Elements, MGE):用于携带各种基因,赋予宿主对抗生素或金属的抗性、致病性、共生性和新底物的代谢能力。
● 代谢通路数据库(Metabolic Pathways From all Domains of Life, MetaCyc):包含参与初级和次级代谢的途径。
● 群体感应数据库(Quorum Sensing Database, QSDB):信号分子的积累使单个细胞能够感知细菌数量的现象,从而使细菌细胞之间能够协调反应和行为。
● 毒力因子数据库(Virulence Factors Database, VFDB):获取细菌毒力因子。
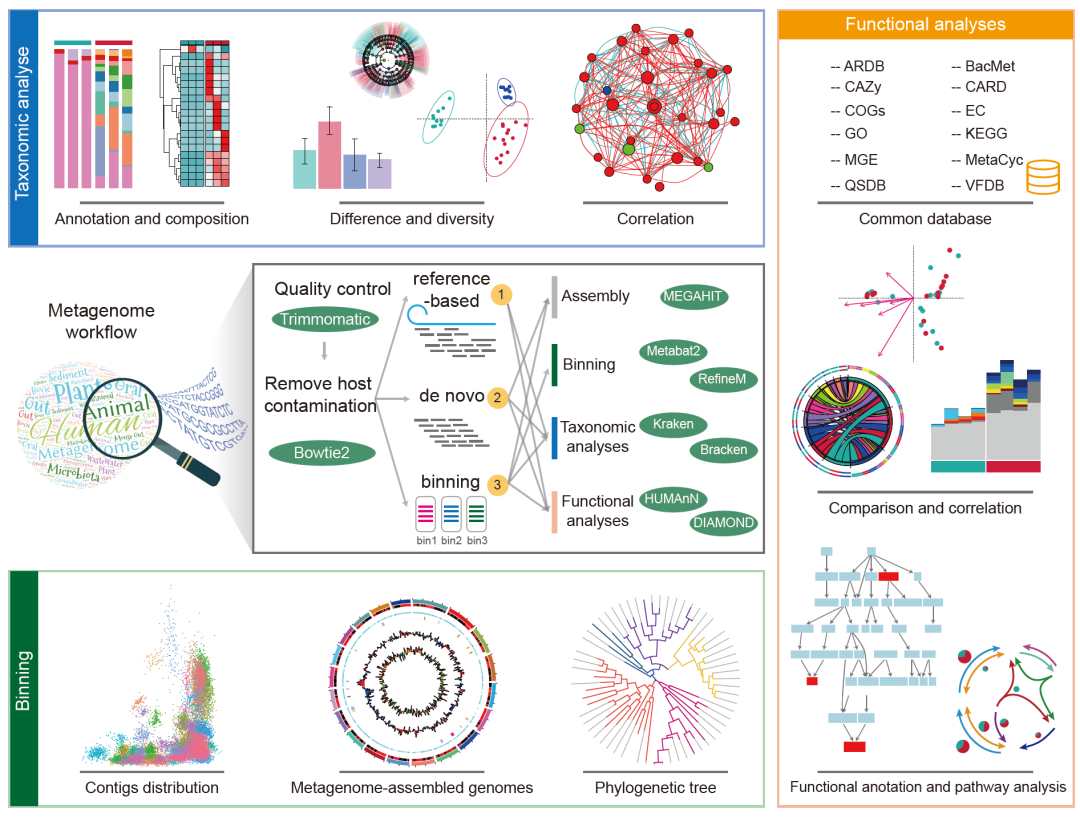
图3. 使用Wekemo Bioincloud平台开展宏基因组数据分析
过滤后的数据(clean data)可以采用有参(reference-based)、组装(de novo)和分箱(binning)的不同流程进行分析,图中简介了组装、分箱、物种和功能分类过程中使用的主要软件和可视化结果。
宏基因组有参云流程是读序(reads)映射到不同的数据库(包括基因、核苷酸或蛋白质序列)。在该流程中,Kraken用于物种分类,Bracken用于估计物种或属水平的丰度。为了呈现物种的注释或组成,可以使用条形图、热图和维恩图;组或样本之间的比较可以使用ANOVA、DESeq2、Kruskal-Wallis和LEfSe;群体的多样性可以通过Bray-Curtis、Bray-Curtis PCoA和α多样性分析来可视化;可以使用热图、网络和冗余分析/典型对应分析来探索组之间或不同因素之间的相关性。此外,使用HUMAnN和UniRef90完整库将质控后读序分配给微生物(采用CARD、COG、KEGG、MetaCyc、GO、EC和CAZy数据库),明确其代谢途径和功能。然后,功能分析的注释或组成、比较或相关性可以像物种分类一样显示。此外,使用DiTing可以分析元素循环(碳、氮、磷、硫)并创建循环路径,用户也可以选择差异基因,比较它们的差异或功能途径。
宏基因组组装云流程,不依赖现有的参考序列,而是生成组装的重叠群,从而有助于发现更多新物种。在这个过程中,物种分类和分析的方法与有参云流程描述类似。而功能分析则是基于组装的重叠群,通过MEGAHIT进行组装,Prodigal进行基因预测。在此过程中,CARD、COG、KEGG、GO、CAZy、MEG、ARDB、BacMet和VFDB数据库用于功能注释。用户还可以选择是否统计基因计数,即特定功能类别内的基因数量。
宏基因组分箱云流程利用了样品中重叠群(contigs)的丰度和覆盖度,以及基于相似Kmer频率和GC含量等对重叠群分组。在此流程中,应用MEGAHIT来组装clean reads并产生重叠群。然后,使用MetaBAT对重叠群进行分箱,使用RefineM消除重叠群以去除高污染的重叠群,使用CheckM评估每个bin的完成度和污染度,使用dRep获得非冗余基因。然后,参考上述描述对binning的物种组成和功能的进行分析和可视化。在这个流程中,CAZy、COG、GO、KEGG、VFDB、MGE和CARD数据库被用于功能注释。此外,还可以创建分箱(bin)的宏基因组组装基因组(MAG),以显示有关不同重叠群的染色体方向、GC含量或GC偏斜(GC-skew)等信息。
案例2:代谢组学数据分析流程
代谢组学可大致分为非靶向(nontargeted)代谢组学和靶向(targeted)代谢组学。这里,平台提供了三种云流程:非靶向、靶向和高通量靶向(high‐throughput targeted)(图4)。非靶向代谢组学特点是无偏见,有助于对生物体的代谢物进行全面分析,帮助我们找到一些新颖的生物标志物。靶向代谢组学具有标品,实现靶向代谢物的绝对定量,并且靶向代谢组是可重复的。高通量靶向代谢组学可以快速有效地分析样品中的大量代谢物,有助于更全面地了解靶向代谢谱。然而,只有靶向代谢组学才能实现代谢物的绝对定量,非靶向和高通量靶向代谢组学通常被认为是相对定量。三种不同的代谢组学方法的数据分析大多相似,包括化合物检测、数据预处理、统计分析、特征选择和功能分析。
平台选用了MetaboAnalystR用于分析非靶向和靶向代谢组学数据,以便在平台中检测代谢物化合物。
● 数据预处理,平台采用质量评估和质量控制以检测异常值,并去除超出三倍标准误差的代谢物或样本。然后,用户可以选择是否进行数据标准化,以消除样本采集或代谢物检测过程中潜在的系统偏差。
● 之后将所有代谢物与KEGG数据库br08001进行比对,确定各生物作用的百分比含量。然后进行标准统计分析,包括化合物总结和结构差异的识别。
● 特征选择,我们提供偏最小二乘判别分析(PLS-DA)或OPLS-DA来区分样本组之间的潜在代谢模式。然后,还采用单变量分析和机器学习技术,例如随机森林和支持向量机,来选择差异代谢物。
● 对于功能分析,我们提供相关分析、通路的过度表征分析(ORA)、通路的拓扑分析和代谢通路图,以直观地了解通路内代谢物的关系、功能模式和拓扑结构。
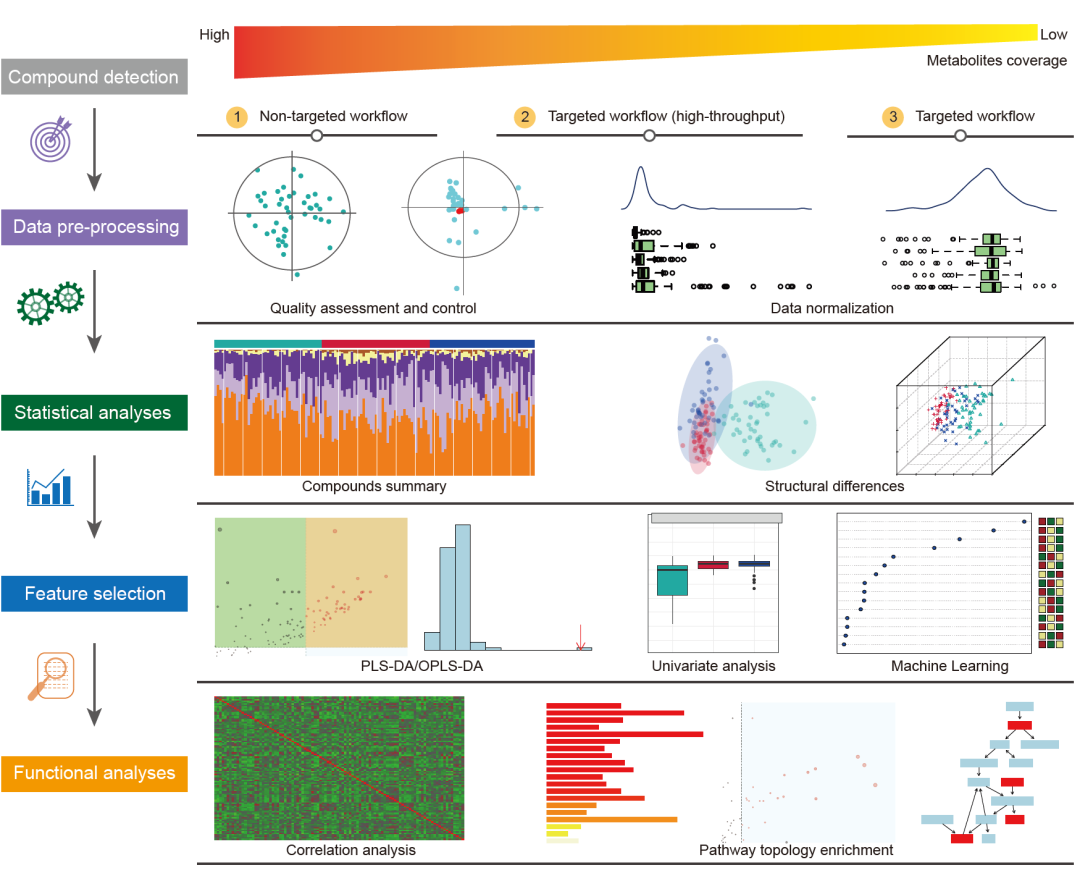
图4. 使用Wekemo Bioincloud平台开展代谢组数据分析
分别针对非靶向(nontargeted)、靶向(targeted)和高通量靶向(high‐throughput targeted)代谢组开发了三种分析流程,主要步骤为数据预处理、常规数据分析、挑选差异代谢物、功能分析。
此外,我们应用maSigPro包来增强非靶向和靶向代谢组云流程的时间序列分析。通过广义线性模型,允许用户识别各种元素(如基因、代谢物或特征)和时间因素(如时间、时间平方或指定组)之间的显著回归关系。此外,平台还构建了一个全面的高通量靶向代谢数据库供用户使用(含超过2500种植物代谢物和约1800种动物代谢物)。
方 法
Wekemo Bioincloud被设计为一个Web应用程序,采用Javascript、HTML、Vue和Bootstrap进行前端开发。对于后端数据预处理和分析,它结合了各种广泛使用的组学分析软件/工具。这些步骤包括但不限于数据质控、去除宿主基因污染、过滤rRNA读序和嵌合体、去冗余、分箱、组装、物种或基因注释、物种/基因/代谢物/蛋白质的定量,功能预测或注释,以及多样性、差异、相关性和系统发育分析。
有关平台使用的所有软件/工具的详细信息,请访问我们的网站 https://www.bioincloud.tech/ 。在这里,我们简要概述了宏基因组有参云流程的步骤。包括Trimmomatic用于FASTQ数据的质量控制、读序过滤和碱基校正;Bowtie将过滤的读序与宿主参考基因组进行比对,以消除宿主DNA污染;Kraken用于物种丰度和分类,HUMAnN用于识别微生物功能,带有 .qzv文件的输出文件可以使用QIIME2查看,多数统计分析和可视化均基于R包获取。
讨 论
Wekemo Bioincloud平台提供强大且用户友好的服务,为多组学研究领域的全球合作做出贡献。与现有的组学数据在线分析平台相比,MicrobiomeAnalyst专注于宏基因组分析,MetaboAnalystR专注于代谢组学分析,GeNets则致力于基因组分析。我们的平台因提供了22个云流程,涵盖扩增子测序数据、宏基因组数据、宏转录组数据、宏病毒组数据、宏蛋白质组数据、基因组数据和理化数据的分析。这种广泛云流程促进了多组学数据的综合分析,为研究人员提供了全面且便捷的解决方案。
同时,Wekemo Bioincloud平台还为研究人员提供了一些个性化的分析,以适应他们多样化的研究范围。例如,我们提供全面的生物信息学数据库(ARDB、BacMet、CAZy、CARD、COGs、EC、GO、KEGG、MGE、MetaCyc、QS、VFDB),用于宏基因组数据的深入功能分析(图3)。该平台支持不同群体肠型的比较,以及样品中蛋白质或原噬菌体分泌的预测(图1)。据我们所知,尽管Majorbio Cloud也是一个用于多组学分析的生物信息学平台,Wekemo Bioincloud平台还提供65种工具(图2)和在线SVG编辑器,使研究人员能够在线美化其图片达到发表需求。
未来,我们打算将EasyMicrobiome和EasyMetagenome流程集成到我们的平台中,并在半年内不断更新平台。虽然该平台目前只有中文版本,但是可通过谷歌翻译进行英文转换,未来我们计划更新英文版本,并增加英语视频教程,以提高国际研究人员的访问和使用。
引文格式:
Gao, Yunyun, Guoxing Zhang, Shunyao Jiang, and Yong‐Xin Liu. 2024. “Wekemo Bioincloud: A User‐friendly Platform for Meta‐omics Data Analyses.” iMeta. e175. https://doi.org/10.1002/imt2.175
作者简介

高云云(第一作者)
● 中国农业科学院深圳农业基因组研究所博士后,2022年博士毕业于北京林业大学。
● 目前研究方向为宏基因组方法开发,相关成果已发表于iMeta、Protein & Cell等期刊。

张国兴
● 2018年研究生毕业于中国农业大学,此后一直在深圳微科盟科技集团有限公司工作,负责生科云的开发。

江舜尧
● 深圳微科盟科技集团有限公司创始人。
● 2011年研究生毕业于深圳大学,先后在华大基因、美吉生物等单位任职,于2017年创立微科盟集团,2019年统筹组织开发生科云,系互联网+科研的开拓者和数智化+科研的探路者。

刘永鑫(通讯作者)
● 中国农科院深圳基因组所研究员,iMeta期刊执行主编,宏基因组公众号创始人。
● 主要研究方向为微生物组方法开发、功能挖掘和科学传播,在Science、iMeta、Nature Biotechnology、Nature Microbiology等期刊发表论文50余篇,被引16000+次,入选全球Top 2%高被引科学家。主编《微生物组实验手册》专著,由300多位同行参与,共同打造本领域长期更新的中文百科全书。创办宏基因组公众号,16万+同行关注,累计阅读量超4千万,打造本领域最具影响的科学传播平台。为Nature Communications、Microbiome、ISME、NAR等68种期刊审稿190次。2022年联合发起iMeta期刊(ESCI收录),打造微生物组/生物信息领域国际顶刊,解决我国本领域期刊出版卡脖子问题,建立国际学术话语权体系,2024年6月即将获得本刊首个影响因子,预计IF~20。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、Scopus等数据库收录。
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 3963
3963

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









