







遗传学和表观遗传学之间的复杂关系是人类健康和疾病的基础。实验和计算能力的进步已经从生物系统的研究中产生了各种各样的高通量数据。大量的技术努力致力于增加吞吐量,并提高实验和计算效率。因此,人们对可以整合不同类型的组学数据并进行关联分析以识别重要参与者和机制的计算方法和软件有明显的兴趣。
2024年3月13日,俄勒冈州立大学Andrey Morgun教授团队在 Nature Protocols 上发表了题为 Transkingdom Network Analysis (TkNA): a systems framework for inferring causal factors underlying host–microbiota and other multi-omic interactions 的方法学论文。研究所提出的跨界网络分析(TkNA)是一种独特的分析框架,通过整合来自多个队列和多种组学类型的数据,推断主宿–微生物群和其他多组学相互作用的因果因素。与大多数只找到关联的多组学方法不同,TkNA专注于在考虑多组学数据的复杂结构的同时建立因果关系,而无需大量的样本大小。

研究提出了跨界网络分析(TkNA),这是一个独特的因果推断分析框架,通过整合来自多个队列和多种组学类型的数据,提供了生物系统的全局视图。TkNA有助于解码在特定条件或疾病中主宿–微生物群(或任何多组学数据)相互作用的关键参与者和机制。TkNA重构了一个网络,该网络代表了捕获生物系统中不同组学之间复杂关系的统计模型。它识别出在多个队列中方向变化和相关性符号的稳健和可重复的模式,以选择差异特征及其每组相关性。然后,该框架使用因果敏感的度量、统计阈值和拓扑标准来确定形成跨界网络的最终边缘。通过随后网络的拓扑特征,TkNA识别控制给定子网络或管理王国和/或子网络之间通信的节点。在TkNA中,进行网络重构所需的数百万个相关性的计算时间通常只需要几分钟,具体取决于研究设计。与大多数只找到关联的多组学方法不同,TkNA专注于在考虑多组学数据的复杂结构的同时建立因果关系。它实现了这一点,而不需要大量的样本大小。此外,TkNA协议用户友好,只需要最小的安装和对Unix的基本熟悉。研究人员可以在 https://github.com/CAnBioNet/TkNA/ 访问TkNA软件。
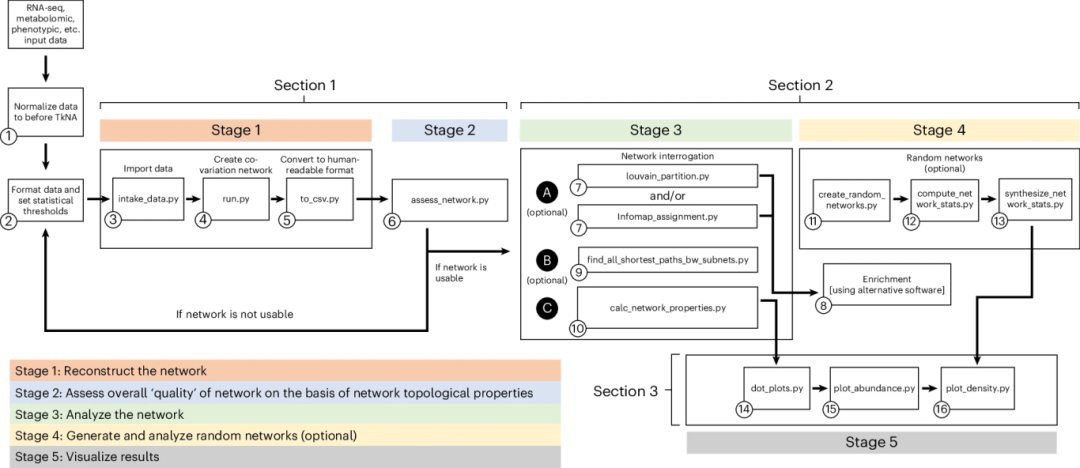
图 1: TkNA的流程图。
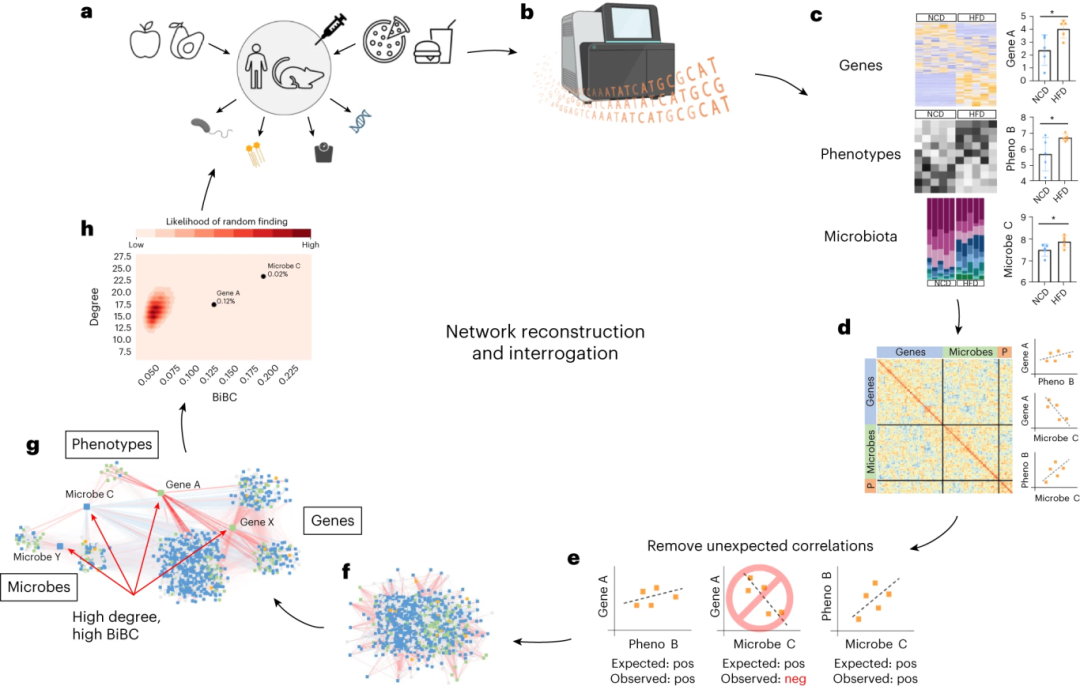
图 2: TkNA流程以循环的方式使用。a,进行实验并获得样本。b,在开始TkNA之前,对样本进行测序和标准化。c,对每种数据类型进行比较。条形图分别代表基因表达水平、微生物丰度和表型的测量水平(均值 ± 标准差)。星号代表处理组之间的显著差异,因为只有显著改变的特征(基于用户定义的统计阈值)才保留用于下游分析。d,执行每种数据类型之间和之内的相关性。e,移除意外的相关性。f,重构网络。g,查询网络以找到调控节点。h,确定从网络中找到顶部节点的概率,然后进行后续验证研究并重复循环。
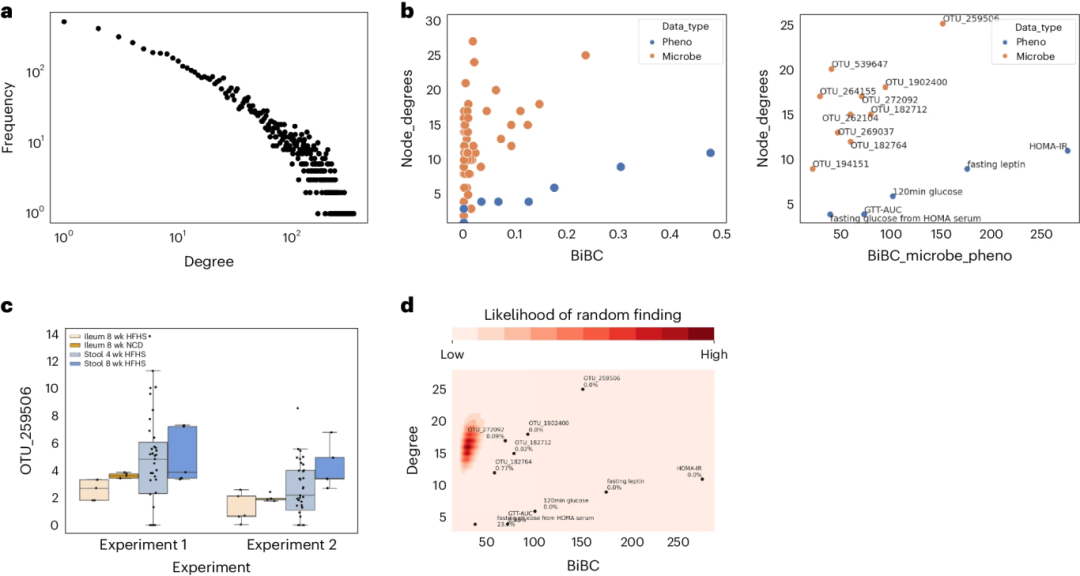
图 3: TkNA生成的示例输出图。a,度分布图。b,左:节点属性可视化示例,其中每个点代表重构网络中的一个节点。右:同一图,放大到前10个微生物BiBC节点。c,b中找到的顶部BiBC节点的丰度/表达图示例。图例显示了数据集中的两个类别。在这种情况下,一个名为“高”的样本类别与一个名为“低”的类别进行了比较。框显示每个实验每组的四分位数;须包括除异常点外的分布的其余部分。d,10,000个随机网络的2D密度图,其中叠加了b中的顶部节点。














 2275
2275











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









