






点击蓝字 关注我们
比较多组学分析揭示品种对猪结肠宿主-微生物相互作用的影响

研究论文
● 原文链接DOI: https://doi.org/10.1002/imo2.8
●2024年7月4日,浙江大学宗鑫团队在iMetaOmics在线发表了题为“Comparative multiomics analyses reveal the breed effect on the colonic host–microbe interactions in pig”的文章。
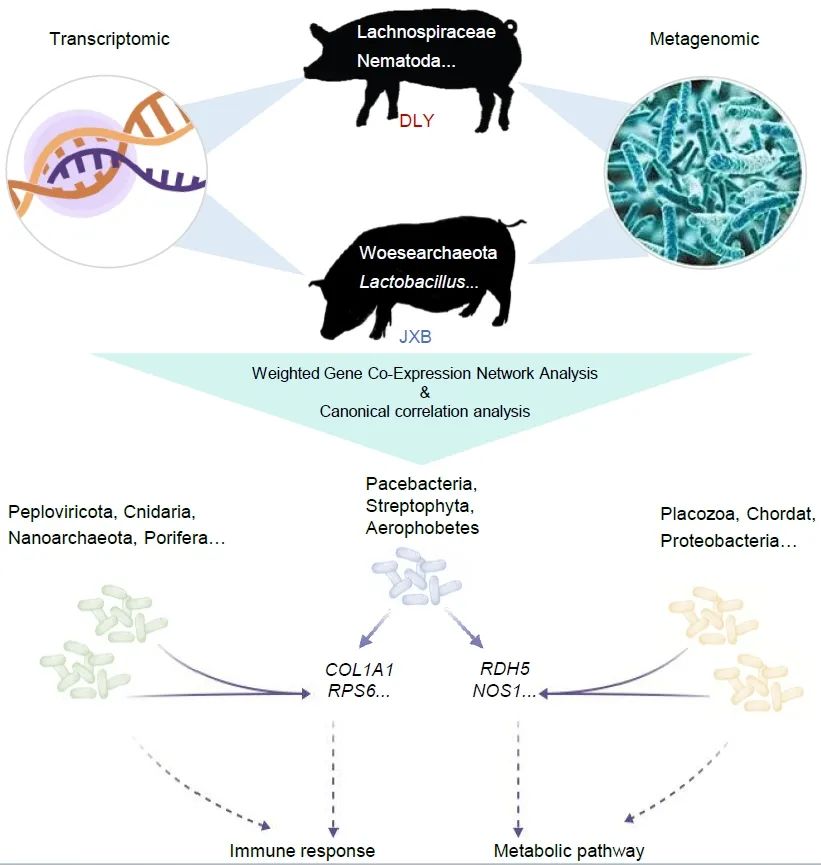
● 本研究揭示了嘉兴黑猪(JXB)和杜洛克×长×大(DLY)猪两个猪品种中肠道微生物群对宿主基因的共同调控和特定调控。
● 第一作者:黄亮、罗世琪
● 通讯作者:宗鑫(zongxin@zju.edu.cn)
● 合作作者:刘舒琦、靳明亮、汪以真
● 主要单位:浙江大学动物科学学院动物分子营养学教育部重点实验室、浙江大学动物科学学院农业部东部动物营养与饲料重点实验
亮 点
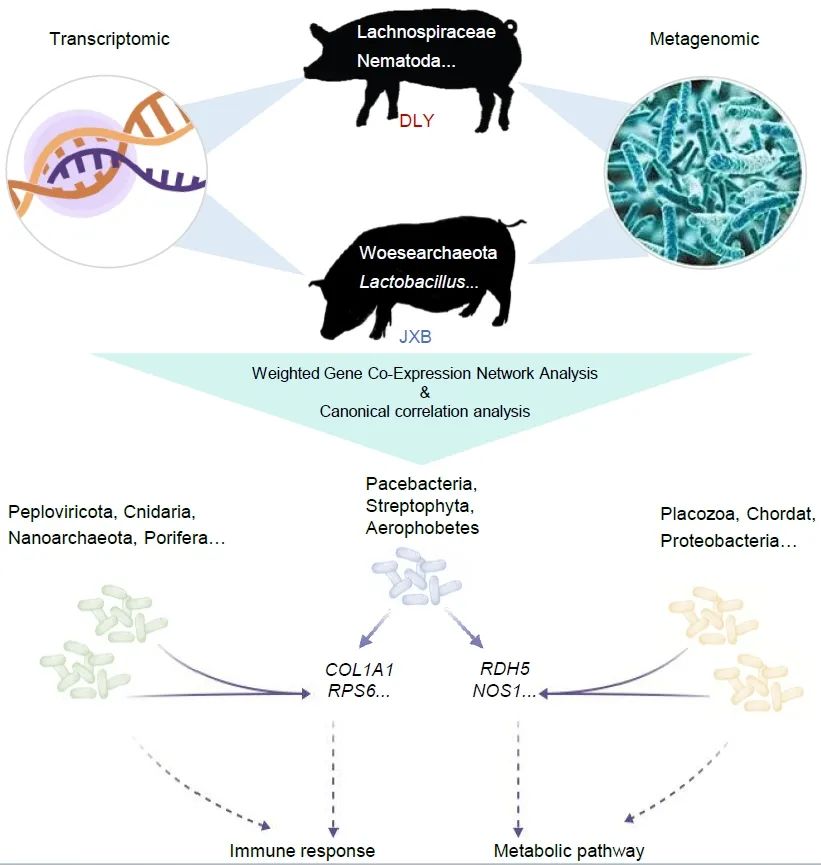
● JXB和DLY的结肠基因表达特征在代谢和炎症反应中高度富集;
● DLY中优势菌群主要为乳酸菌,而JXB中毛螺杆菌科为主要的优势菌群。此外,Pacebacteria、Streptophyta和Aerophobetes被鉴定为与PI3K-Akt介导的免疫应答相关的关键细菌;
● 含Ig样结构域蛋白参与调节肠道菌群代谢,而ITIH2、PAEP和TDRD9参与NLR介导的肠道菌群免疫应答。
摘 要
肠道菌群失调常导致免疫相关疾病、消化不良或腹泻。本研究以中国本地猪品种嘉兴黑猪(JXB)为研究对象,对其进行宏基因组和转录组综合分析,揭示其肠道微生物群基因和肠道组织基因表达通路的相互作用。在JXB猪和杜洛克×长×大(DLY)猪之间共发现452个差异表达基因(DEGs)和174个细菌门。进一步分析发现,JXB和DLY在结肠基因表达特征上的差异主要富集在代谢和炎症反应上,其中乳酸菌和毛螺杆菌科分别在DLY和JXB中富集。值得注意的是,在两种猪品种中均发现Pacebacteria、Streptophyta和Aerophobetes参与了PI3K-Akt介导的免疫反应;此外它们只在JXB猪中促进其肠道代谢。相对,宿主可通过Ig样结构域蛋白、ITIH2、PAEP和TDRD9等基因调节微生物代谢和免疫应答。综上所述,我们的研究结果揭示了两个猪品种中肠道微生物群对宿主基因的共同调控和特定调控。
视频解读
Bilibili:https://www.bilibili.com/video/BV181421b78t/
Youtube:https://youtu.be/FFisU-yICII
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
肠道微生物群在宿主体内与其共同处于动态平衡,但其组成和功能在个体之间差异很大,并对宿主代谢、免疫反应和其他关键生理途径起关键调节作用。肠道菌群稳态与宿主疾病密切相关,尤其是代谢性疾病。肠道菌群失调通常会导致炎症性肠病(IBD)和结肠炎等疾病,这对于个性化药物治疗非常重要。
肠道微生物群的结构可以通过外部刺激调节,如饮食、抗生素治疗和母体微生物群。因此,研究人员提出假设,宿主的基因型可以影响其微生物群的组成,作为反馈微生物群的组成又反过来影响宿主的表型。因此,检测它们的相互作用以揭示它们在人类肠道疾病发病机制或动物生产中的作用至关重要。杜×长×大(DLY)三元杂交猪是在全球范围内通过先进育种选择的商品猪品种,在不影响抗病性的情况下实现快速生长。相对地,嘉兴黑猪(JXB)是中国太湖地区的一个地方猪品种,具有性成熟早、繁殖能力强、耐粗饲料、抗病等特点,但生长、瘦弱、屠宰率较低。这二者不同的性状为研究品种介导的微生物-宿主相互作用提供了理想的动物模型。考虑到结肠及其内容物具有更长的运输时间和更丰富的细菌群的特点,我们选择了这部分肠道进行研究。
为了研究结肠细菌对猪宿主的贡献,我们重点研究了JXB和DLY猪的结肠微生物群与宿主基因的相互作用。我们收集了190日龄的DLY和JXB猪(各5头)的结肠组织和结肠内容物,进行了RNA-seq和宏基因组测序。我们确定了肠道微生物与宿主基因(涉及宿主代谢和免疫反应)之间的相互作用,从而深入了解了品种对宿主-微生物组相互作用的影响。
结 果
两个不同品种的结肠微生物群的微生物组成
为了检测肠道微生物的不同组成,我们对两个猪种(DLY和JXB)的宏基因组数据进行了统计分析,在门水平上共检出208个类群(表S1)。Shannon和Simpson指数显示,这两个猪品种之间的肠道菌群多样性和丰度没有显著差异(图1A, B)。主坐标分析(PCoA)也显示出相似的beta多样性(图1C)。但偏最小二乘-判别分析(PLS-DA)显示两组之间存在差异(图1D)。拟杆菌门(Bacteroidetes)与厚壁菌门(Firmicutes)的相对丰度比在JXB中升高(图1E、F)。使用线性判别分析效应量(LEfSe)来鉴定优势微生物类群,我们发现DLY中优势菌群主要为乳酸菌(Lactobacillus),而毛螺杆菌科(Lachnospiraceae)在JXB中富集(图1G和表S2)。
为了进一步了解这两个品种的肠道菌群结构,我们进行了微生物共现网络分析。与JXB肠道相比,DLY肠道中结肠微生物之间的相关性在属水平上更广泛(图1H)。值得注意的是,JXB的肠道菌群表现出更大的模块化系数(0.885 vs. 0.76)和特征向量中心度(0.02968 vs. 0.01984),表明虽然它们相互作用的计数更少,但相互关系更紧密。
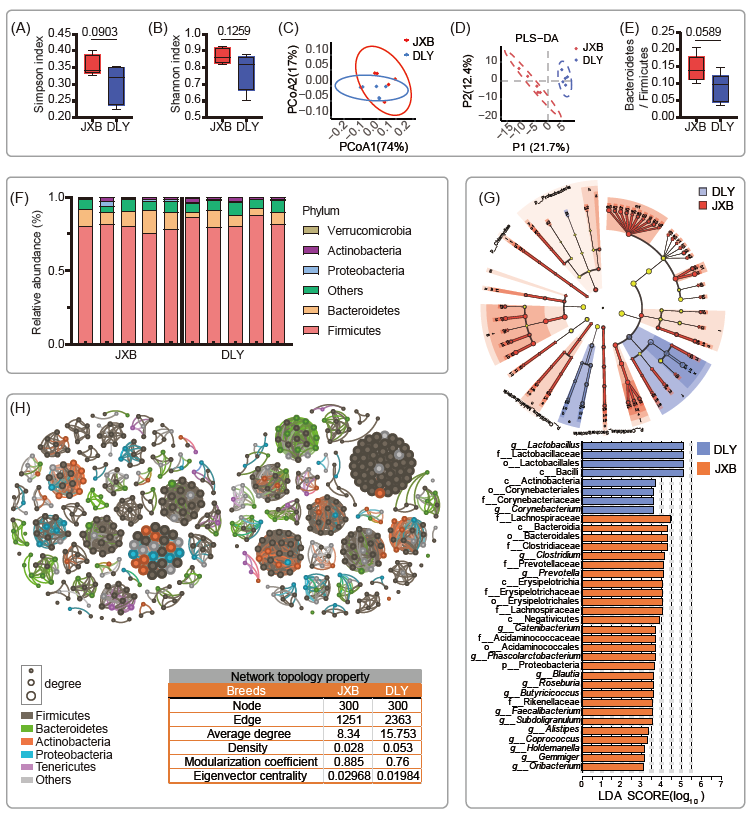
图1. 嘉兴黑猪(JXB)和杜洛克×长×大猪(DLY)的肠道细菌组成(每组N = 5)
(A)两个猪种之间α多样性的差异由Shannon指数和(B)Simpson指数表示;(C)主坐标分析(PCoA)和(D)偏最小二乘判别分析(PLS-DA)显示了两组之间的差异;(E)两组中拟杆菌门与厚壁菌门的比例;数据以均数±均数的标准误差(SEMs)表示;(F)堆叠图表展示门水平肠道菌群相对丰度;(G)利用线性判别分析效应量(LEfSe)分析两组群落组成差异;图中不同颜色的节点表示对组间差异有显著影响,黄色节点表示未发生显著变化的微生物类群;列显示LDA > 3的生物标志物;(H)两组中微生物共现的视觉网络和拓扑统计;LDA:线性判别分析效应量。
耐药基因组是微生物组的一个分支,由抗生素耐药基因(ARG)组成,危害着人类的健康。我们研究了这两个品种之间肠道耐药基因组的差异。我们在两个品种中鉴定出304个ARG,它们之间的α或β多样性无显著差异(图S1A-C)。比较前10个抗生素ARG的相对丰度,发现大环内酯类和头孢菌素类抗生素基因在JXB肠道中显著上调,而链霉素和链菌素-a则下调(图S1D)。为了揭示两个品种的ARG的组成结构,我们构建了两个品种的ARG的微生物共生网络,并表明杆菌肽在两个品种的ARG中发挥了核心作用(图S1E, F)。进一步通过随机森林分析确定了贡献最大前30个的ARG,其受试者工作特征(Receiver Operating Characteristic, ROC)曲线下的面积为1 (p = 0.0625,准确率优于“无信息率”的95%置信区间)(图S1G-I)。
DLY猪和JXB猪结肠基因表达谱的比较
为了检测转录水平的差异,我们进行了RNA-seq。对表达水平(rpm)高于0.001的基因进行了计数,共检测到24,212个基因(图S2A和表S3)。在JXB和DLY中分别检测到1104和1223个基因。主成分分析(PCA)显示两组之间的距离较小(p = 0.4415 (PC1)和0.6471 (PC2))(图S2B)。为了识别二者的差异,我们将差异倍数> 2且p-adjust < 0.05的基因识别为差异表达基因(DEGs)。由此在JXB品种中,检测到233个基因上调,219个基因下调(图S2C, D)。
我们进一步通过Kyoto Encyclopedia of Genes and Genomes(KEGG)数据库进行富集分析以确定基因功能的差异。结果表明,与DLY猪相比,JXB猪上调的基因主要富集在代谢通路(图2A, B)。此外,JXB猪中上调的基因也在核苷酸代谢、精氨酸和脯氨酸代谢等通路中富集。JXB猪下调的基因主要富集在昼夜节律、ABC转运蛋白、细胞色素P450代谢外源物质等通路上。整合所有DEGs进行富集分析,发现代谢通路和细胞因子-细胞因子受体相互作用通路显著富集,强调了这两个猪种结肠组织中代谢和炎症反应的差异。为了揭示DEGs和通路之间的关系,我们构建了所有DEGs的蛋白相互作用网络,但在蛋白水平上未观察到这些通路之间有直接联系(图S2E)。综上所述,JXB猪的代谢水平和免疫应答与DLY猪不同,代谢水平升高可能是由于代谢通路相关基因表达上调所致。
为了确定导致JXB和DLY结肠差异的基因,我们进行了加权基因共表达网络分析(WGCNA)。最终,452个DEGs被分为12个模块(图2C, D)。模块2、4、8、9、11和12聚类,并被确定为JXB标志性模块。相反,模块1、5、6、7和10聚集在DLY结肠中(图2E, F)。值得注意的是,模块6和模块11分别与JXB和DLY猪呈现出最强的正相关,因而认为这两个模块中的基因对JXB猪和DLY猪结肠之间的差异起着至关重要的作用。随后,我们找出了模块6和模块11中的关键基因, IQCF5(ENSSSCG00000025367)和靶向RNA的DNA聚合酶(ENSSSCG00000048052)(图2G)。
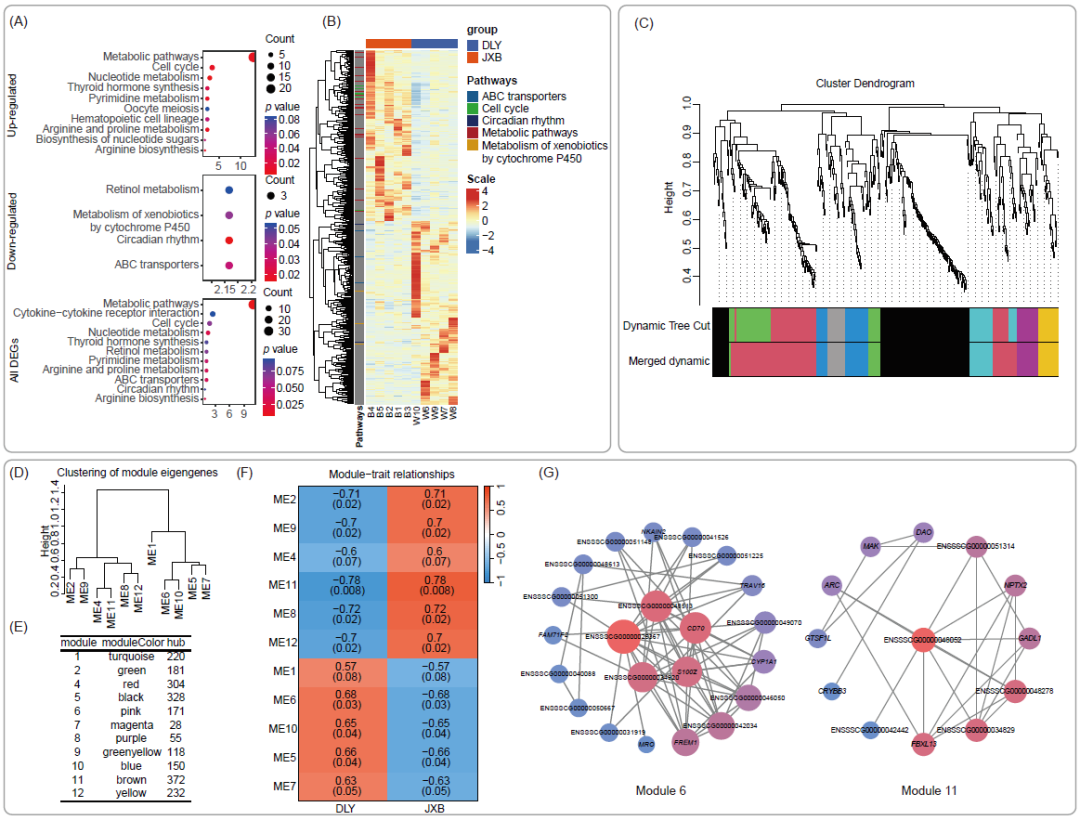
图2. 嘉兴黑猪(JXB)和杜洛克×长×大猪(DLY)(每组5只)基因集的转录变化
(A)气泡图显示差异表达基因(DEGs)富集的通路;(B)热图显示了DEGs的表达,它们所属的通路也被标记;(C)簇树图;(D)各模块基因计数;(E)模块的进化树;(F)通过加权基因共表达网络分析(WGCNA)发现物种和模块之间的相关性;不同颜色代表表达模式相似的基因组成的不同模块,相关系数下方显示p值;(G)网络显示模块的关键驱动,节点的梯度颜色和大小表示度(基因的边数),红色表示高度,蓝色表示低度。
宿主途径和肠道微生物群之间的相互作用
近年来,典型相关分析(CCA)已成为分析微生物-宿主相互作用的常用工具。为了解究竟哪些微生物类群参与调控JXB和DLY猪肠道中的差异通路,我们整合10头猪(每个品种5头)的数据进一步展开了稀疏CCA分析,将转录组中的所有基因分为上调基因(Up)、下调基因(Down)和无差异基因(Common)进行分析。Common组基因从表达量最高、方差最低的前25%基因中选取,共4674个基因。随后,我们获得了与这些基因组相互作用的不同微生物群集(表S4-S6)。
将各组中与菌群存在互作的基因用于KEGG富集分析,结果显示Common组的基因在参与免疫应答和代谢调节的多条通路中富集(图3A),我们展示了其中富集基因数量最多的免疫、代谢相关通路基因组成及其对应的相互作用微生物(图3B)。Up组基因主要富集在代谢通路,通过与结肠的基因(如LOC100522404、RDH5、NOS1)相互作用,肠道微生物可以增强嘉兴黑猪的代谢(图3C)。Pacebacteria、Streptohyta和Aerobbeta与PI3K-Akt信号通路和代谢通路均存在相互作用,表明它们在嘉兴黑猪的肠道中促进和维持其代谢和免疫。
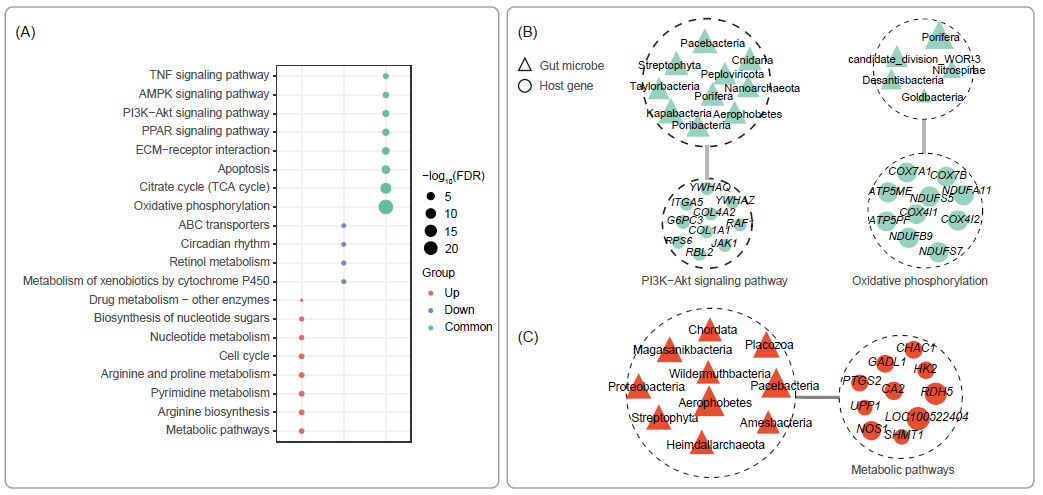
图3. 嘉兴黑猪(JXB)和杜洛克×长白猪(DLY)的宿主途径-肠道菌群相互作用(每个基因集N = 10)
(A)气泡图显示了与富集基因相关的Kyoto Encyclopedia of Genes and Genomes(KEGG)通路;通过稀疏典型相关分析(CCA) (FDR < 0.85)筛选与肠道菌群的交互作用基因;(B)肠道菌群和Common组宿主基因(在两个品种中表达没有变化的基因)的关联;三角形和节点大小分别代表稀疏CCA系数基因和微生物的绝对值;图中颜色代表各组,红色代表Up组基因(JXB猪上调基因),绿色代表Common组基因;(C) Up组肠道菌群与宿主基因的关联。
为了研究JXB猪和DLY猪中DEGs和肠道菌群之间的差异相互作用,我们对每个猪品种的452个DEGs和肠道菌群进行了相关性分析,并构建了它们的相互作用网络,相关性|cor| > 0.4, p < 0.05被视为微生物-基因相互作用(表S7-S8)(图S3A)。基于这些数据,我们确定了以品种依赖方式形成的相互作用(图S3A,左和右),以及与交集基因发生相互作用的不同微生物类群(图S3A,下)。在参与相互作用的微生物中,有150个交集菌群(图S3B)。我们从中确定了对这两个品种的特定DEGs发挥调节作用的微生物类群(图S3B,左和右),以及与交集微生物相互作用的DEGs(图S3B,下)。这些结果展示了两个猪品种中差异丰度的细菌和宿主基因之间相互作用的异同。
宿主基因对肠道菌群的调节
为了检验宿主基因对肠道菌群的影响,我们通过与KEGG数据库比较分析了肠道菌群的功能。我们观察到,JXB猪的肠道微生物在NOD样受体(NLR)介导的免疫应答中表现更好,因为其宏基因组reads在NOD样受体信号通路中富集(图4A)。相应地,DLY微生物组中富集的通路主要与代谢相关,尤其是甘油脂代谢。此外,在DLY猪中,缬氨酸、亮氨酸和异亮氨酸的降解途径以及酮体的合成和降解途径普遍存在,这表明该品种的饲料转化率提高可归因于其肠道菌群。
随后,我们在NOD样受体信号通路和甘油脂代谢中确定了显著改变的子集KEGG Orthology (KO),并利用它们确定了相关的DEG模块(图4B)。我们特意从NOD样受体信号通路中选择K02406,从甘油脂代谢中选择K00086、K11529、K00128和K00865,从代谢通路中选择K07676、K21089、K11936和K11936进行后续相关性分析(图4C),因为这些KO的reads计数变化最显著。数据显示模块5与甘油脂代谢和代谢途径相关的各种KOs具有显著的相关性(图4D)。同样,我们发现模块7和模块8与NOD样受体信号通路中的K02406密切相关。因此,我们构建了一个相关网络来识别这些模块中的关键基因。结果显示免疫球蛋白样结构域蛋白(ENSSSCG00000034961)是调节DLY肠道菌群代谢功能的关键宿主基因,而ITIH2、PAEP和TDRD9主要参与调节JXB肠道菌群NLR介导的免疫应答。这些结果为宿主基因如何调节肠道微生物提供了一个很好的例子。
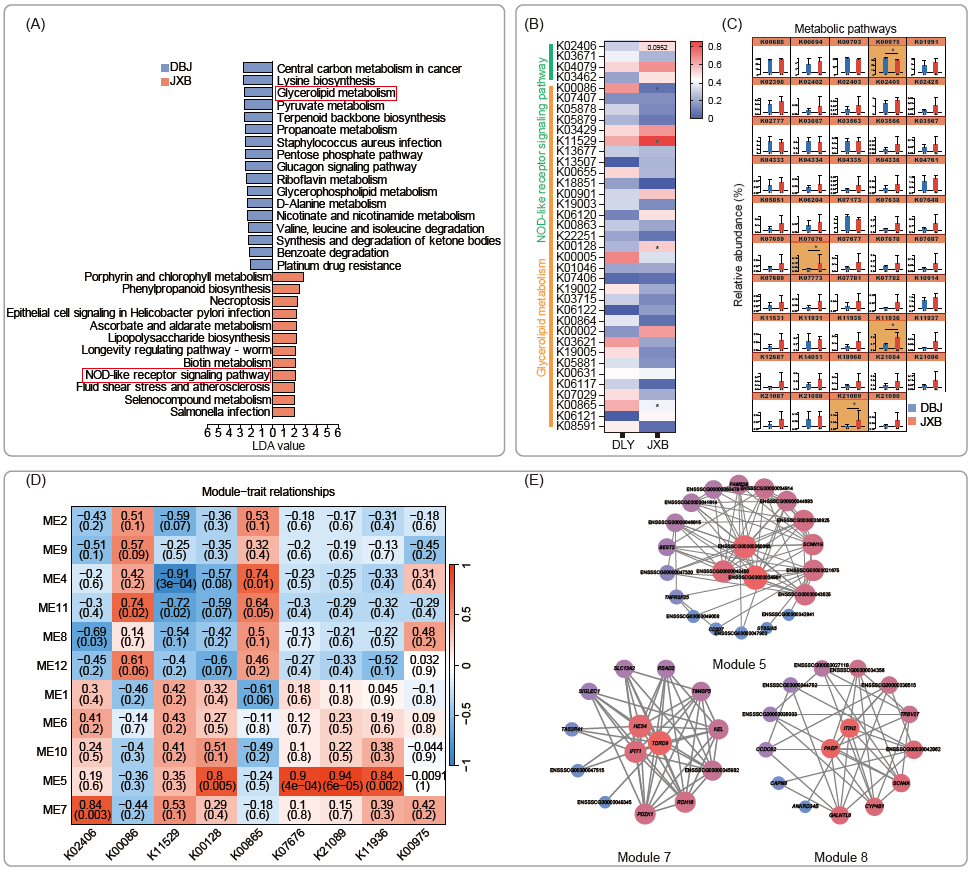
图4. 嘉兴黑猪(JXB)和杜洛克×长×大猪(DLY)肠道菌群功能差异(每组N = 5)
(A)每个品种的Kyoto Encyclopedia of Genes and Genomes Orthology(KO)的线性判别分析效应量(LEfSe);(B)热图显示了在NOD样受体信号通路和甘油脂代谢中KO的不同读片计数,通过Mann-Whitney检验,*表示p < 0.05的显著变化;(C)柱状图显示参与代谢通路的KO的reads计数;(D)物种和模块之间的相关性,每个单元中存在两个值代表相关系数(上)和p值(下);(E) Network表示模块的关键驱动,节点的梯度颜色和大小表示度(基因的边数),红色表示高度,蓝色表示低度。
方 法
收集嘉兴黑猪(JXB)和杜洛克×长白猪(DLY)的结肠及其内容物进行转录组学和宏基因组学分析。通过加权基因共表达网络分析和稀疏典型相关分析,我们识别了宿主基因与肠道微生物之间的潜在相互作用,参与PI3K-Akt信号通路和代谢通路的宿主基因受到肠道微生物的调控。
讨 论
对JXB猪的研究主要集中在肉质,导致了一些研究致力于对其肌肉的检测,目前尚未存在该品种的结肠组织相关研究。以Yorkshire猪和Min猪为模型,多篇报道表明,宿主-微生物群交互作用促进了疾病抗性表型。因此,我们探讨了JXB中发生的交互作用类型及其如何影响宿主表型。在本研究中,我们主要关注了两个品种中与免疫和代谢相关的肠道微生物-宿主相互作用的差异,同时也描述了两个品种中均存在的相互作用(如图3中的Common组)。
在DLY猪品种中检测到的较高丰度的Woesearchaeota和Lactobacillus导致了它们更强的消化和吸收率。研究发现,JXB猪种中含有较多的微生物,如Saccharibacteria、Nematodes和Melainabacteria,尤其是Lachnospiraceae和Bacteroidia,这使得其群落结构更活跃,抗氧化应激反应能力更强,对粗饲料的耐受性更强。
与PI3K-Akt信号通路正相关的菌群在JXB结肠内容物中更丰富。此外,变形菌门(Proteobacteria)在JXB猪中富集,而据报道,变形菌门可以影响宿主血清IgA库,并对细菌性脓毒症提供结构性保护。Placozoa有许多免疫相关基因,并参与共生菌群和抵抗外源性病原体,研究显示它与JXB猪肠道的代谢途径存在相互作用。这些发现意味着该品种的肠道免疫应答能力更强。包含Ig样结构域的蛋白参与免疫应答,有趣的是,我们发现它也调节肠道微生物的代谢,这意味着包含Ig样结构域的蛋白作为枢纽将细菌代谢和宿主免疫联系了起来。但目前还没有相关的研究报告,需要进一步的研究来证实。
宿主基因和微生物群落之间不同的相互作用模式为这两个猪品种之间生产力差异的潜在机制提供了初步的见解。这些发现也可为了解肠道菌群的作用机制及其在畜牧业生产中的潜在应用提供参考。本研究仍存在诸多不足,需要更深入的实验验证来探究肠道菌群与宿主基因之间的相互影响和因果关系。
代码和数据可用性
本研究中使用的所有原始数据均可在NCBI SRA数据集中获得(BioProject: PRJNA1005787,网址:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1005787/)。使用的数据和脚本保存在https://github.com/hillincre/DLY-and-JXB-multi-omics-analysis。补充材料(表格、图表、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新材料)可以在在线DOI或iMetaOmics Science http://www.imeta.science/imetaomics/上找到。
引文格式:
Liang Huang, Shiqi Luo, Shuqi Liu, Mingliang Jin, Yizhen Wang, Xin Zong. 2024. Comparative multiomics analyses reveal the breed effect on the colonic host–microbe interactions in pig. iMetaOmics 1: e8. https://doi.org/10.1002/imo2.8
作者简介

黄亮(第一作者)
● 浙江大学动物科学专业博士,硕士毕业于广西大学。
● 研究方向主要为脂代谢和肠道菌群,相关学术成果发表于International Journal of Biological Sciences、 iMetaomices等期刊。

罗世琪(第一作者)
● 浙江大学动物科学饲料与动物营养方向硕士。
● 本科就读于华南农业大学动物科学学院,后考入浙江大学攻读硕士学位,在浙江大学良渚实验室交流学习。目前的研究方向主要是炎症下的翻译组变化。

宗鑫(通讯作者)
● 浙江大学研究员,博士生导师。
● 围绕断奶仔猪肠道免疫和脂肪酸吸收开展研究,在猪肠道健康营养调控、改善断奶仔猪腹泻等方面取得多项创新性研究成果,以第一作者/通讯作者(含共同)在Nucleic Acids Research、Journal of Advanced Research等国内外主流期刊发表论文20余篇,授权发明专利5项;主持国家自然科学基金面上项目、青年基金、国家重点研发子课题等。
iMetaOmics
相关资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 757
757

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









