






点击蓝字 关注我们
TCfinder:基于通路活性在单细胞RNA数据中鉴别肿瘤细胞

方法论文
● 原文链接DOI: https://doi.org/10.1002/imo2.22
● 2024年8月7日,上海科技大学刘雪松团队在iMetaOmics在线发表了题为“TCfinder: Robust tumor cell discrimination in scRNA-seq based on gene pathway activity”的文章。
● 本研究开发了TCfinder (Tumor Cell finder),一种基于通路得分的肿瘤细胞识别工具,它摆脱了传统方法对特定标记基因的依赖,并克服了单细胞数据稀疏的问题。
● 第一作者:吴宸旭、宁伟
● 通讯作者:刘雪松 (liuxs@shanghaitech.edu.cn)
● 合作作者:吴涛、陈静、要慧子、陶紫玉、赵翔宇、刁凯旋、王金禹、王卫良、李新星、宋倩倩
● 主要单位:上海科技大学生命与科学技术学院
亮 点
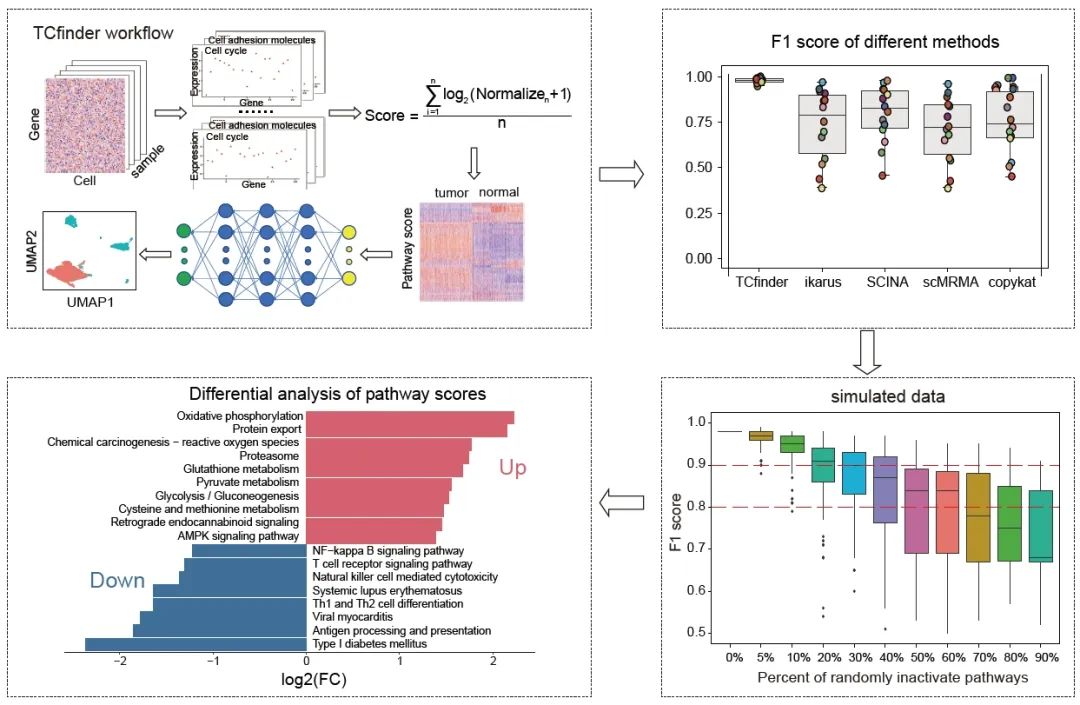
● TCfinder是一个易用的R包,使用通路得分来区分正常细胞和肿瘤细胞;
● TCfinder的肿瘤细胞识别性能优于现有工具;
● TCfinder性能稳定,在稀疏的单细胞数据中依然表现良好。
摘 要
单细胞RNA测序(scRNA-seq)已成为研究肿瘤的重要方式之一,不仅揭示了细胞间相互作用,还提供了对癌症演变和异质性的深入了解。然而,在稀疏的单细胞数据中准确区分肿瘤细胞和正常细胞仍然是一个挑战。在此,我们开发了TCfinder (Tumor Cell finder),一种基于通路得分的肿瘤细胞识别工具,它摆脱了传统方法对特定标记基因的依赖,并克服了单细胞数据稀疏的问题。在多个独立的scRNA-seq数据集中,TCfinder在肿瘤细胞识别方面表现优于现有方法。此外,对TCfinder重要特征的研究中发现,抗原呈递通路在肿瘤细胞中的表达低于正常细胞。总体而言,TCfinder被证明是一种准确的基于通路的泛癌肿瘤细胞与正常细胞鉴别工具。
视频解读
Bilibili:https://www.bilibili.com/video/BV14w4m1k7j9/
Youtube:https://youtu.be/dGVHoIwvsEc
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
传统的肿瘤大块组织RNA测序(RNA-seq)只能获得基因表达的平均信息,因此无法精确描绘肿瘤微环境和浸润细胞的状态。相比之下,单细胞RNA测序(scRNA-seq)技术能够深入了解细胞生态系统,包括对特定细胞群体及其各自转录组特征的研究,展示肿瘤内部异质性,构建肿瘤进化图谱等。
对肿瘤组织scRNA-seq数据中的肿瘤细胞进行分析的主要挑战之一是准确快速地区分肿瘤细胞和正常细胞。虽然许多细胞类型注释方法已经被构建用于scRNA-seq分析,但很少有工具能够用于区分肿瘤细胞和正常细胞。目前,有两种常见策略用于在scRNA-seq数据中区分肿瘤细胞和正常细胞。一种策略是聚类手动注释。虽然手动注释能够有效区分单细胞数据中的肿瘤细胞和正常细胞,但它仍然受到单细胞数据集稀疏性的限制,并且高度依赖专业经验和知识,难以大规模推广。另一种策略是自动注释,包括基于标志基因的方法,如ikarus,SCINA,scMRMA,或基于拷贝数变异(CNV)推断的方法。然而,由于scRNA-seq数据比较稀疏,导致大多数癌细胞的基因标志物表达不足,因此这种策略存在一定限制。此外,CNV在肿瘤细胞中并不普遍存在,一些正常细胞也可能具有CNV,这限制了基于CNV的方法的广泛应用。因此,开发一种能够克服单细胞数据稀疏性、广泛适用的自动注释方法,对于单细胞肿瘤研究领域来说具有重要意义。
由于典型的基因通路通常包含数十个基因,基于通路得分克服了传统基因标志物注释方法所面临的数据稀疏性问题。此外,基因通路的改变是癌细胞和正常细胞之间的主要区别之一。因此,表征基因通路在准确区分癌细胞和正常细胞方面具有巨大潜力。
在此,我们基于通路得分和深度神经网络(DNN)开发了TCfinder。TCfinder不仅在具有随机基因失活的模拟scRNA-seq数据中表现出稳健性能,还在多种癌症类型中与现有方法相比表现出更高的肿瘤与正常细胞区分精度和准确性。TCfinde 软件的最新版本已经在GitHub (https://github.com/XSLiuLab/TCfinder)中发布,软件包中还提供了测试用的演示数据集。
结 果
用于肿瘤细胞识别的通路
我们在京都基因和基因组百科全书(KEGG)数据库中收集了所有的人类基因通路,并对每个通路的活性进行了评分,以获得单细胞通路评分矩阵。在训练数据集中,我们对肿瘤细胞和正常细胞之间的通路评分进行了Wilcoxon检验,保留了p < 0.05的通路。最终,我们确定了在TCfinder中使用的213条通路(表S1)。在TCfinder中,我们利用一个完全连接的神经网络架构开发了一个区分肿瘤细胞和正常细胞的模型(图1A)。为了开发一个广泛适用于泛癌症水平的模型,我们从六种类型的癌症中收集了7万多个细胞作为训练数据。这些数据集以8:2的比例被随机分为训练集和测试集。此外,我们使用了包含超过23万个细胞的10个数据集作为模型的独立验证数据(表S2)。
比较TCfinder与现有的注释方法
利用独立数据集比较了TCfinder与现有ikarus、SCINA、copykat、scMRMA等方法的性能。TCfinder在10X建库的数据集上平均F1得分为0.98,在SMART-Seq2建库的数据集上F1得分为0.95(表S3)。其他四种方法的性能与TCfinder相比F1评分、准确性和精度都较低(图1B,S2)。由于肿瘤和正常细胞的金标准scRNA-seq数据集相当有限,为了解决这一问题,我们使用健康组织样本作为正常样本,而使用肿瘤细胞系作为肿瘤样本。我们使用了GSE162616数据集的健康个体的单细胞数据和GSE140440数据集的肿瘤细胞系进行比较检测。与现有方法相比,TCfinder的假阳性率和假阴性率更低(图1C,S3)。
对识别肿瘤和正常细胞重要的通路
使用limma包对肿瘤细胞和正常细胞的通路评分进行差异分析,|log2FC|> 1且FDR< 0.05的通路作为差异通路(图1D)。结果表明,肿瘤细胞中活跃的途径主要与代谢有关,如氧化磷酸化、活性氧、谷胱甘肽代谢和糖酵解/糖异生等。相反,在肿瘤细胞中被抑制的途径主要与免疫功能有关,包括抗原加工和呈递、Th1和Th2细胞分化,以及自然杀伤细胞介导的细胞毒性。这些发现表明,从单细胞的角度来看,肿瘤细胞表现出代谢过度活跃和免疫抑制的双重特征。
为了进一步研究哪些通路对肿瘤细胞的识别贡献最大,我们对每个通路进行了随机打乱,并计算了模型精度的差异,这可以反映该通路对模型的重要性。经过100次随机分组,我们发现四个最重要的途径有助于肿瘤细胞的识别:I型糖尿病、氧化磷酸化、病毒性心肌炎和抗原加工和呈递(图1E)。为了验证这些通路是否在肿瘤组织测序中也很突出,我们收集了癌症基因组图谱(TCGA)的数据。结果显示,除了氧化磷酸化途径外,其他三个与自身免疫和抗原呈递相关的途径在肿瘤样本中并没有出现较高的通路得分,这表明bulk组织不能完全揭示潜在的肿瘤细胞表达差异(图S5)。我们进一步研究了这些贡献通路中的共享抗原呈递(AP)相关基因,这些基因包括MHC I和MHC II等基因(图S6)。这些AP相关基因出现在单细胞肿瘤细胞中,而不是在bulk组织中(图S7),这表明一些关键的肿瘤细胞相关信息在bulk组织RNA-Seq中被掩盖了。
讨 论
我们开发了TCfinder,这是一种新的基于基因通路评分的深度学习方法,可以准确、快速地区分scRNA-seq中的癌细胞和正常细胞。当只有少量的基因在单细胞中被测到时,通路得分值将反映整个通路的活动,这克服了单细胞数据的稀疏性问题。在多个独立的数据集和模拟数据中,TCfinder的性能比现有的方法有所提高。尽管TCfinder已经证明了它在多个单细胞数据集上克服数据稀疏性的能力,并显示出了良好的性能,但它仍然有一些局限性。其中一个主要的限制是用于训练模型的注释单细胞数据集数量较少。尽管TCfinder有其局限性,但在解决单细胞注释中的数据稀疏性问题方面是对现有方法的一个重大改进。它在这方面的成功也可能为注释其他细胞类型提供有用的见解。
代码和数据可用性
本研究仅使用了公开的数据,其数据来源和处理见材料、方法和表S2。TCfinder是一个R软件包,可以在GitHub( https://github.com/XSLiuLab/TCfinder)安装和使用。复现本文结果所需的代码可以在:https://github.com/XSLiuLab/TCfinder/tree/master/inst/analysis.上获得
引文格式:
Chenxu Wu, Wei Ning, Tao Wu, Jing Chen, Huizi Yao, Ziyu Tao, Xiangyu Zhao, Kaixuan Diao, Jinyu Wang, Weiliang Wang, Xinxing Li, Qianqian Song, Xue-Song Liu. 2024. "TCfinder: Robust tumor cell discrimination in scRNA-seq based on gene pathway activity." iMetaOmics: e22. https://doi.org/10.1002/imo2.22
作者简介

吴宸旭(第一作者)
● 上海科技大学2022级博士研究生。
● 目前研究方向为肿瘤基因组变异,多组学数据挖掘,深度学习。

宁伟(第一作者)
● 上海科技大学2020级硕士研究生。
● 主要研究方向为组学数据挖掘。

刘雪松(通讯作者)
● 上海科技大学生命学院研究员、博士生导师、教授。
● 研究方向聚焦肿瘤基因组及肿瘤精准诊断治疗。近 5 年在知名学术期刊(Cancer Research, eLife,Brief Bioinform,Oncogene) 上发表通讯作者研究论文 20 多篇,多篇通讯作者研究论文被ESI标记为“高被引论文”,连续入选 2022、2023 年度 Elsevier 发布的“中国高被引学者”榜单。著有《癌症生物学》教科书。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
●iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
更多推荐
(▼ 点击跳转)
iMeta | 引用13000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,定位IF>10的高水平综合期刊,欢迎投稿!
联系我们
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 123
123

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









