






 点击蓝字↑↑↑“微生态”,轻松关注不迷路
点击蓝字↑↑↑“微生态”,轻松关注不迷路
编译:微科盟Sky蓝,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
肠道细菌与炎症性肠病 (IBD) 有关,但有关菌株尚不清楚。大规模的微生物组进化研究可以揭示疾病在肠道菌群上的印记,从而确定可能导致炎症的菌株和基因。在这里,我们使用数千名 IBD 患者和健康对照者的粪便宏基因组数据重建了 14万种菌株基因型,揭示了数百种在IBD中富集的谱系。我们证明这些菌株是古老的,分类学上是多样的,并且在人体中普遍存在。此外,在炎症期间,与疾病相关的菌株比健康的菌株更有竞争力,这意味着对疾病的长期适应。菌株遗传差异映射到已知的炎症轴上,包括氧化应激、营养生物合成和免疫逃避。最后,与健康相关的迟缓艾格特菌(Eggerthella lenta )菌株的消失可预测粪便钙卫蛋白,这是一种疾病严重程度的生物标志物。我们的工作确定了可能影响炎症疾病的菌株多样性库,并可以扩展到其他微生物相关的疾病。
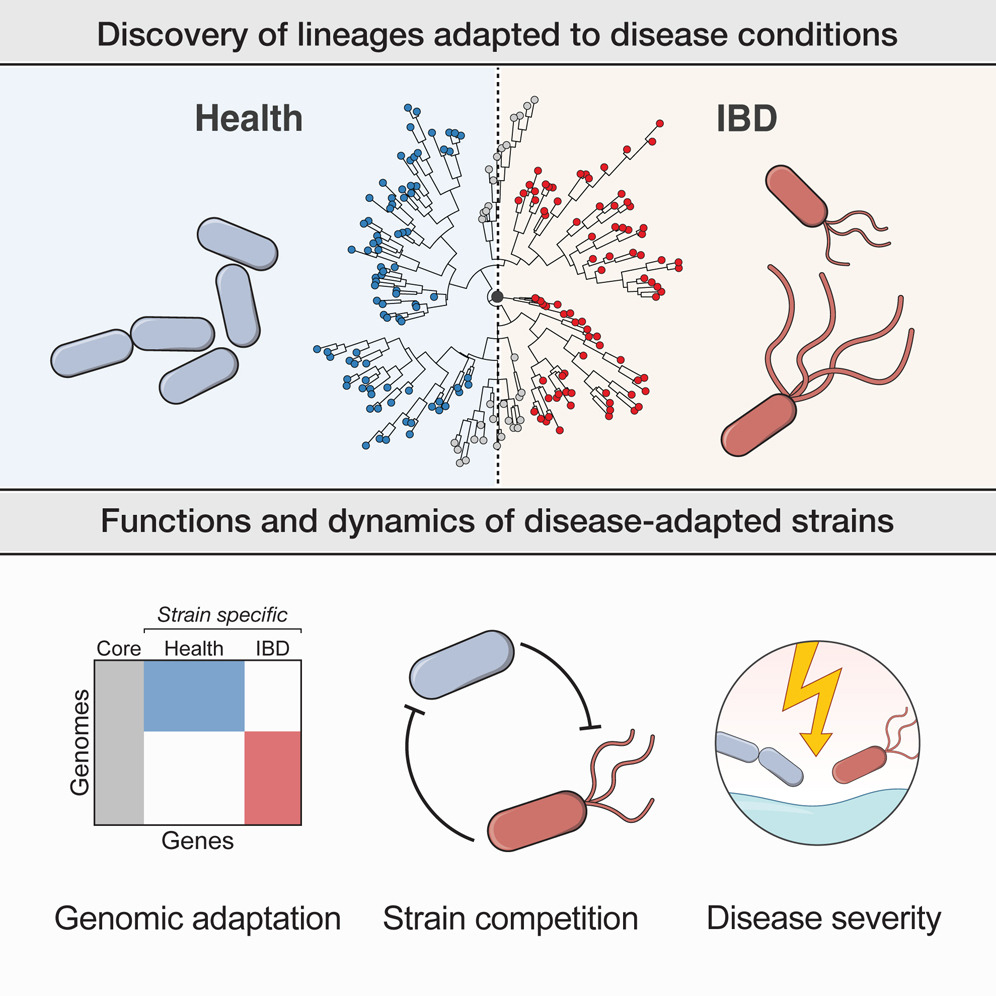
图文摘要
论文ID
原名:Discovery of disease-adapted bacterial lineages in inflammatory bowel diseases
译名:炎症性肠病中疾病适应细菌谱系的发现
期刊:Cell Host & Microbe
IF:20.6
发表时间:2024.6
通讯作者:Christopher S. Smillie
通讯作者单位:美国麻省总医院
DOI号:10.1016/j.chom.2024.05.022
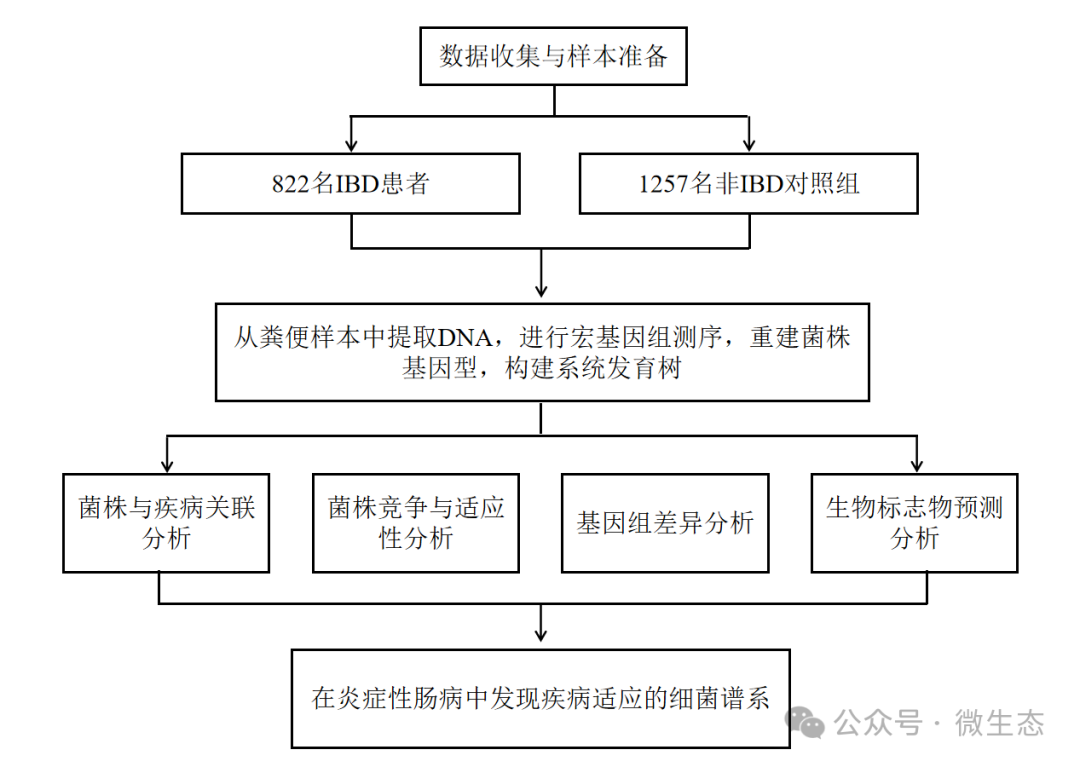
实验设计
结果
1 细菌多样性与IBD相关
为了探索与IBD相关的细菌的遗传多样性,我们首先在所有公共基因组的元数据中搜索与IBD相关的关键词(STAR方法)。在RefSeq数据库中超过50万个基因组中,只有不到500个是从患有IBD的个体中分离或衍生出来的(图1B,S1D和S1F)。尽管宏基因组分析生成了额外的数据,但这些数据集产生的短读长尚未被用于研究细菌的进化。因此,我们对IBD和其他炎症性疾病中细菌多样性和进化的了解极为有限。
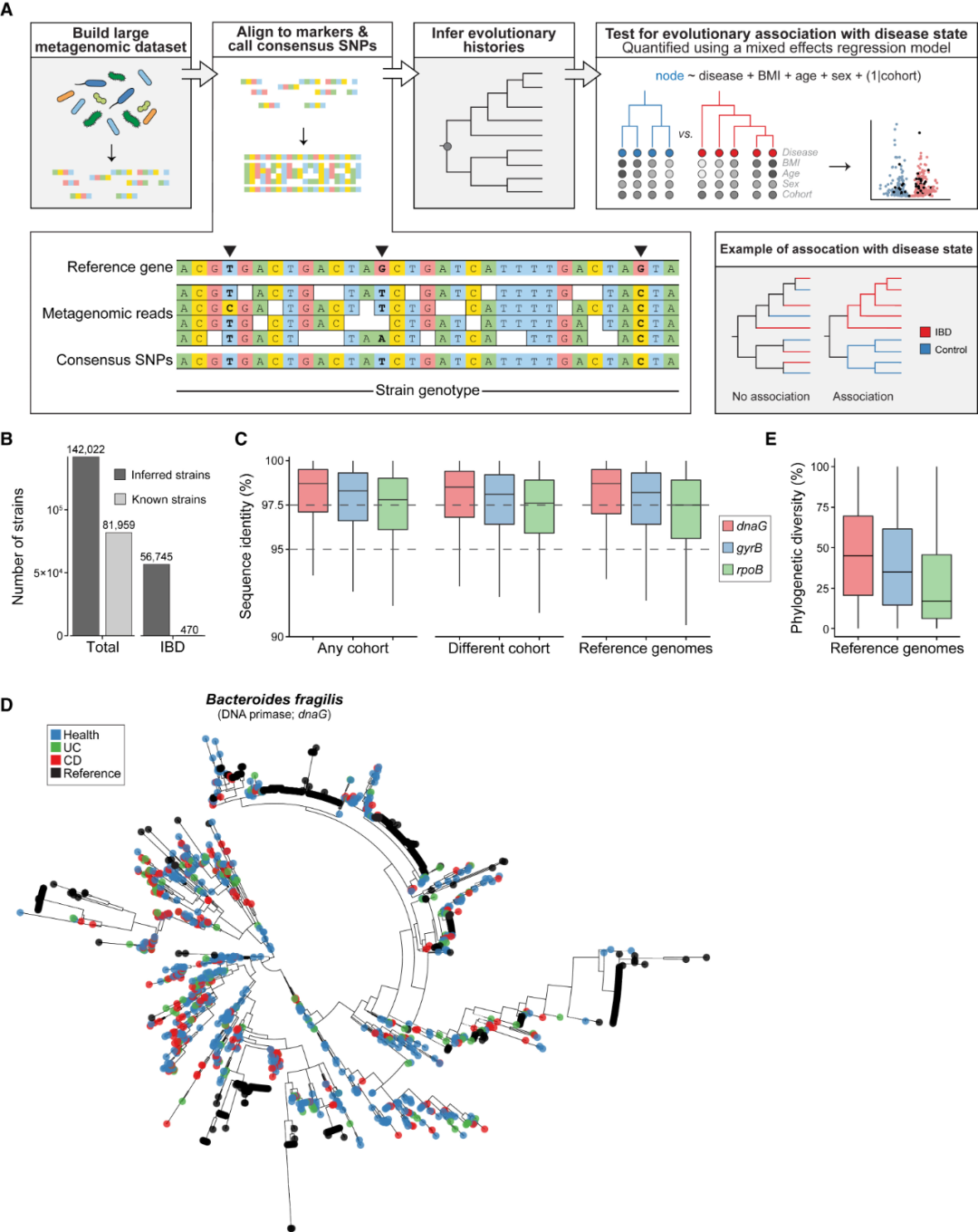 图1. 推断的菌株基因型是IBD中细菌多样性的未探索的储存库。(A)发现疾病相关菌株的工作流程。宏基因组读取与参考菌株标记基因对齐。每个样本中的优势菌株基因型根据对齐位置的一致核苷酸确定。构建了系统发育树,然后测试每个分支在控制其他协变量的情况下在健康或疾病中的富集情况。(B)推断的菌株扩展了已知的疾病衍生细菌。在所有物种中,推断出的菌株(深灰色;基于3基因组)或参考基因组(浅灰色)的基因型数量(y轴);每个条形图顶部显示了总基因型数量。(C)菌株推断的可重复性。对于dnaG、gyrB和rpoB菌株基因型(彩色图例),序列相似性的箱线图(y轴)与以下组中最接近的基因型(x轴,从左到右):任何研究中的菌株;不同研究中的菌株;和参考基因组。(D)代表性菌株系统发育。B. fragilis中dnaG的最大似然系统发育树,显示了推断出的菌株基因型(红色:克罗恩病[CD],绿色:溃疡性结肠炎[UC],蓝色:非IBD)与参考dnaG序列(黑色)的整合。(E)推断的基因型代表未被探索的多样性。对于dnaG、gyrB和rpoB菌株基因型(x轴;彩色图例),箱线图显示了参考基因组捕获的总系统发育多样性的比例(即,共享的分支长度;y轴)。(C和E)箱线图:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。
图1. 推断的菌株基因型是IBD中细菌多样性的未探索的储存库。(A)发现疾病相关菌株的工作流程。宏基因组读取与参考菌株标记基因对齐。每个样本中的优势菌株基因型根据对齐位置的一致核苷酸确定。构建了系统发育树,然后测试每个分支在控制其他协变量的情况下在健康或疾病中的富集情况。(B)推断的菌株扩展了已知的疾病衍生细菌。在所有物种中,推断出的菌株(深灰色;基于3基因组)或参考基因组(浅灰色)的基因型数量(y轴);每个条形图顶部显示了总基因型数量。(C)菌株推断的可重复性。对于dnaG、gyrB和rpoB菌株基因型(彩色图例),序列相似性的箱线图(y轴)与以下组中最接近的基因型(x轴,从左到右):任何研究中的菌株;不同研究中的菌株;和参考基因组。(D)代表性菌株系统发育。B. fragilis中dnaG的最大似然系统发育树,显示了推断出的菌株基因型(红色:克罗恩病[CD],绿色:溃疡性结肠炎[UC],蓝色:非IBD)与参考dnaG序列(黑色)的整合。(E)推断的基因型代表未被探索的多样性。对于dnaG、gyrB和rpoB菌株基因型(x轴;彩色图例),箱线图显示了参考基因组捕获的总系统发育多样性的比例(即,共享的分支长度;y轴)。(C和E)箱线图:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。
为了解决这一重大缺口,我们设想可以从大型IBD宏基因组学数据中重新构建菌株基因型。我们从马萨诸塞州总医院炎症性肠病研究前瞻性登记(PRISM)队列中443名个体的粪便样本中测序了超过960个宏基因组,并将其与公共数据集结合起来,构建了一个包含来自822名IBD患者(515名克罗恩病患者,307名溃疡性结肠炎患者)和1,257名非IBD(名义上‘健康’)对照组个体的6,138个粪便宏基因组的庞大资源(表S1)。这些数据代表了2009年至2021年间收集的11项研究,涵盖了广泛的人口统计学和临床特征,包括疾病状态(健康、克罗恩病、溃疡性结肠炎)、年龄、体重指数(BMI)和性别(图S1A)。对于一些受试者,我们还有纵向测量数据,我们最初进行了子采样以确保统计独立性(STAR方法)。正如预期,基于相对物种丰度的无监督样本聚类将样本按健康状态和队列进行了分组。
为了从这些数据中推断出数十万个高置信度的菌株基因型,我们将所有6,138个宏基因组与肠道中所有已知细菌物种的基因目录(人类胃肠道基因组[UHGG])进行了比对。为了确保准确的系统发育重建,我们使用了来自AMPHORA基因目录的31个系统发育标记基因,我们还补充了DNA旋转酶(gyrB)基因。由于这些基因是垂直遗传的、高度保守的,并且在所有细胞中普遍存在,我们有很强的先验知识认为它们存在于与健康和疾病相关的细菌中。相比之下,其他菌株推断方法使用的基因集通常是由测序的参考基因组确定的,这些基因集往往偏向于健康相关菌株中的基因。相比之下,其他菌株推断方法所使用的基因集通常是根据测序的参考基因组确定的,这些基因集往往偏向于健康相关菌株中的基因。在我们最初的进化分析中,我们使用了一组三个已建立的菌株标记基因,这些基因与多种蛋白复合体相关:DNA引物酶(dnaG)、DNA旋转酶(gyrB)和RNA聚合酶(rpoB)。尽管GyrB和RpoB是环丙沙星和利福昔明的靶标,但DnaG不是抗生素的靶标。我们使用所有32个标记基因验证了我们的主要发现。总的来说,我们生成了数千个比对,每个比对都得到了数百名受试者和多项研究的支持(3基因比对:152名受试者,6项研究;32基因比对:362名受试者,8项研究)。
接下来,为了从这些比对中的短读长中估计高置信度的菌株基因型,我们使用了一致的单核苷酸多态性(SNP)作为主导菌株基因型(图1A;STAR方法)。这是一种经过广泛验证的方法,已被其他菌株推断方法广泛使用。尽管其他方法可以恢复丰度较低的菌株,但我们选择保守地只使用每个样本中最稳健的菌株基因型进行系统发育推断。这种方法与推断更复杂的菌株混合物的方法相比有以下几个优点:(1)通过一致的核苷酸推断基因型不需要强有力的先验假设;(2)主导菌株对测序错误具有鲁棒性,因为它们是由多个reads支持的;(3)菌株在统计上是独立的,因为每个样本只估计一种菌株。相比之下,稀有菌株在统计上依赖于同一样本中估计的其他菌株,这使得下游的统计分析变得复杂。
使用这些方法,我们推断了超过65万个单基因菌株基因型,每个菌株在50 %的比对中支持17个reads的平均读取深度(中位数12.27 , Q1 = 8.9 , Q3 = 19.2)。我们验证了我们推断的基因型在独立样本、不同受试者(通常来自不同的研究)以及“金标准”参考基因组序列中可重复发现。首先,一个人体内估计的菌株基因型通常与其他人甚至其他研究中独立估计的菌株基因型相匹配(图1C;所有菌株的相似性中位数为97.5%;另见图S1G)。其次,推断出的菌株基因型平均与参考序列有超过95%的相似性,且往往完全相同(图1C)。最后,这些金标准参考序列在菌株系统发育中整合,而不是形成独立的外群(图1E和S1E)。例如,脆弱拟杆菌(B. fragilis)参考序列在dnaG基因型的菌株系统发育中整合(图1D)。尽管已知的参考序列经常在菌株系统发育中聚集在一起,但这可能反映了细菌分离和培养中众所周知的偏见,即偏向某些谱系。总之,推断的菌株基因型在独立受试者、其他研究以及RefSeq和UHGG数据库中已发表的参考基因组中可重复检测。
为了研究健康和IBD背景下肠道菌群的进化,我们接下来为一组535个去重复的细菌基因组生成了系统发育树,涵盖了360个不同的物种。我们不依赖于单基因比对,这可能反映了基因特异性的选择压力,而是使用了更加稳健的多基因比对,这些比对整合了dnaG、gyrB和rpoB位点。对于这些标记基因多拷贝的参考基因组,我们确定了一组最优的dna G、gyr B和rpo B等位基因,它们在不同样品中的相对丰度最大( STAR Method )。总的来说,这535个三基因系统发育涵盖了142,022个菌株(健康85,277个;IBD衍生56,745个),代表 IBD 中的菌株基因型增加了 100 倍(图1B)。因此,这些 IBD 相关菌株构成了尚未探索的细菌多样性的丰富来源。
2 与健康和IBD相关的菌株谱系在肠道微生物群中普遍存在
接下来,我们研究了与健康或疾病强烈相关的菌株谱系,这可能为了解慢性炎症期间肠道细菌的功能角色提供新的见解。我们设计了一个统计框架,系统地识别与健康或IBD相关的菌株。具体来说,我们在控制其他协变量(如年龄、性别、BMI和队列(即“批次”))的同时,测试菌株系统发育树的每个节点与健康状态的关联(图1A;STAR方法)。尽管某些研究提供了其他临床数据,如炎症严重程度,但在这里我们专注于大多数数据集中共享的数据。关键的是,我们的方法可以识别与健康或疾病进化且无法用其他因素解释的菌株谱系。与 BMI 相关或研究独有的菌株(即“批次效应”)将不会被恢复。
我们广泛验证了这种方法。首先,系统发育测试与基于加权UniFrac距离的健康与IBD衍生菌株之间的度量是一致的(图S2E)。其次,从dnaG、gyrB和rpoB的单基因系统发育中估计的结果相似,并且大体上与从更稳健的三基因系统发育中估计的结果相匹配(图S2D)。因此,我们的结果不是由基因特异性的选择压力驱动的(如抗生素耐药性),而是反映了菌株的系统发育历史。作为阳性对照,我们鉴定了与包括年龄和BMI在内的其他宿主特征显著相关的菌株;然而,作为阴性对照,没有菌株与性别显著相关(图S2G)。
通过将这些方法应用于包含大多数肠道物种的535个参考基因组,我们发现健康和与IBD相关的菌株在肠道微生物群中普遍存在(图2A、2B、2D和S4)。例如,在健康和疾病期间,普氏栖粪杆菌(Faecalibacterium prausnitzii)和肠道拟杆菌(Bacteroides intestinalis)的不同谱系在个体中富集(图2A)。总的来说,535个系统发育中有107个包含了与健康状态显著相关的菌株谱系(图2A和2B)。此外,富集的程度超过了零假设模型(置换测试;图2C)。具有健康和IBD相关菌株的物种在分类学和生态学上是多样化的,涵盖了严格厌氧菌和兼性厌氧菌;糖、黄酮类化合物和粘蛋白的代谢;以及抗生素耐受性。它们还包括许多先前与疾病相关的物种,如F. prausnitzii、迟缓艾格特菌(Eggerthella lenta)和Faecalicatena gnavus,这表明未被探索的谱系可能是这些已知与疾病联系的基础(图S4)。总的来说,这些发现强烈表明,与疾病相关的多种条件决定了这些菌株的进化,而不是单一的选择力量(例如抗生素)。
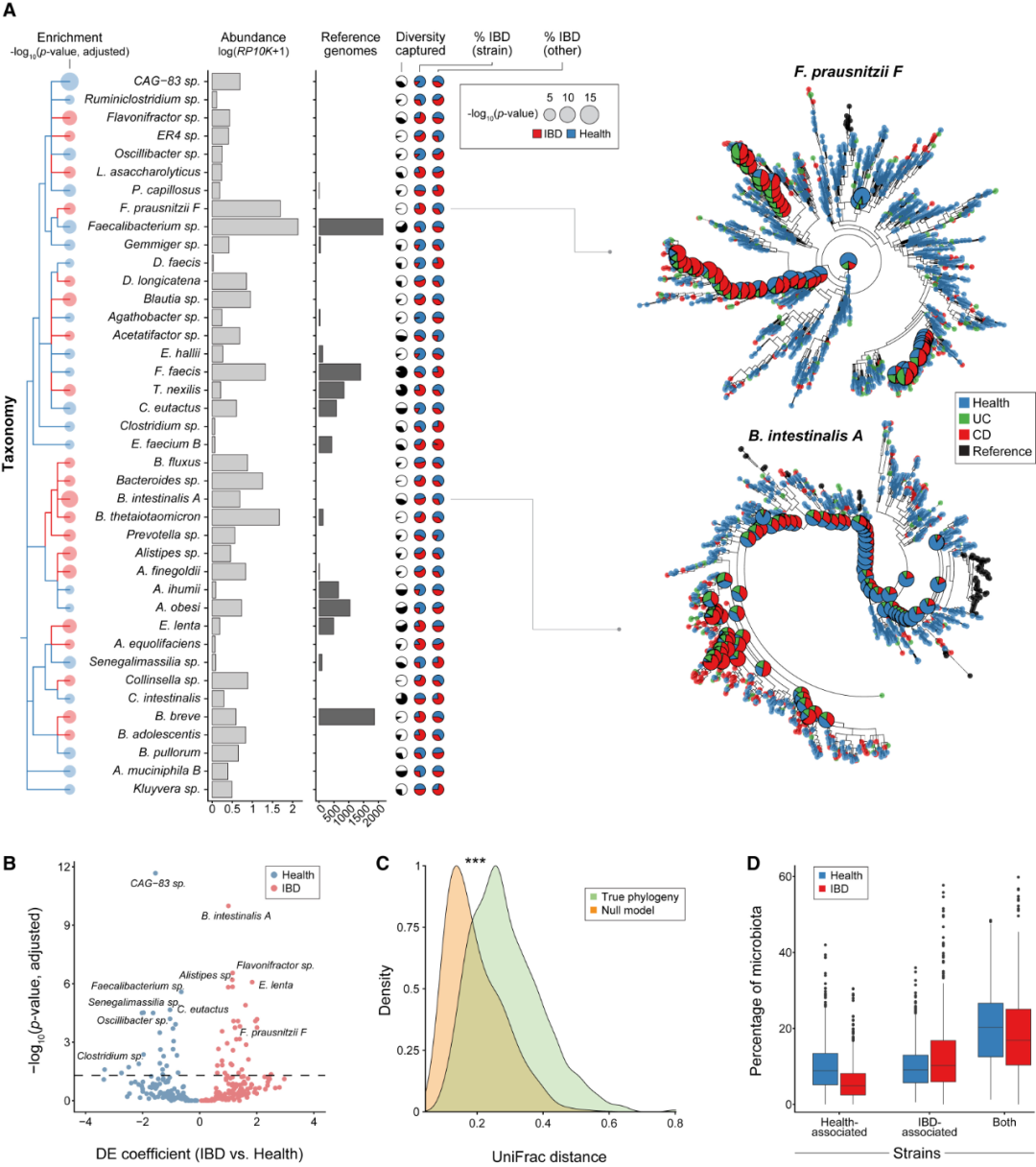 图2. 与健康和IBD相关的菌株谱系在肠道微生物群中普遍存在。(A)健康和IBD相关菌株概述。左侧:菌株系统发育(蓝色:与健康相关,红色:与IBD相关),末端表示最富集菌株的显著性(STAR 方法)。中间:跨菌株,物种的宏基因组相对丰度的平均值(log10(RP10K+1));参考基因组的数量;菌株捕获的系统发育多样性的比例;以及最明显富集菌株和所有其他菌株的疾病状态分布情况。右侧:F. prausnitzii 和 B. intestinalis 的代表性系统发育关系,显示与属于不同谱系的菌株相关的疾病状态。所示为具有3基因组的系统发育树。点(末端):来自健康(蓝色),来自IBD(红色:CD,绿色:UC)。饼图:重要节点的健康状况分布(校正p < 0.05;STAR方法)和所有菌株的分布(根)。(B) 健康和IBD相关的菌株在肠道微生物群中普遍存在。火山图显示了疾病中系统发育富集的效应大小(x轴;混合模型系数;STAR 方法)以及使用 3 基因组(STAR方法)推断的物种系统发育中最显著节点的统计显著性(y 轴)。虚线:校正后的 p = 0.05。(C) 相对于零模型的疾病相关谱系的富集。真实菌株系统发育(绿色)与零模型(橙色)之间的加权标准化UniFrac距离的分布,其中零模型中每个样本相关的健康状况被置换(橙色)。展示了3基因组比对的结果。菌株系统发育包含的UniFrac距离大于零模型(p < 2.2×10−16;Kolmogorov-Smirnov检验),表明健康和疾病之间存在显著的聚集。(D)与疾病相关的菌株在肠道中大量存在。箱线图显示了健康人(蓝色)和IBD患者(红色)的微生物群中由健康相关菌株、IBD相关菌株或这两组菌株组合(x轴)所占的百分比(y轴)。箱线图包括:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。
图2. 与健康和IBD相关的菌株谱系在肠道微生物群中普遍存在。(A)健康和IBD相关菌株概述。左侧:菌株系统发育(蓝色:与健康相关,红色:与IBD相关),末端表示最富集菌株的显著性(STAR 方法)。中间:跨菌株,物种的宏基因组相对丰度的平均值(log10(RP10K+1));参考基因组的数量;菌株捕获的系统发育多样性的比例;以及最明显富集菌株和所有其他菌株的疾病状态分布情况。右侧:F. prausnitzii 和 B. intestinalis 的代表性系统发育关系,显示与属于不同谱系的菌株相关的疾病状态。所示为具有3基因组的系统发育树。点(末端):来自健康(蓝色),来自IBD(红色:CD,绿色:UC)。饼图:重要节点的健康状况分布(校正p < 0.05;STAR方法)和所有菌株的分布(根)。(B) 健康和IBD相关的菌株在肠道微生物群中普遍存在。火山图显示了疾病中系统发育富集的效应大小(x轴;混合模型系数;STAR 方法)以及使用 3 基因组(STAR方法)推断的物种系统发育中最显著节点的统计显著性(y 轴)。虚线:校正后的 p = 0.05。(C) 相对于零模型的疾病相关谱系的富集。真实菌株系统发育(绿色)与零模型(橙色)之间的加权标准化UniFrac距离的分布,其中零模型中每个样本相关的健康状况被置换(橙色)。展示了3基因组比对的结果。菌株系统发育包含的UniFrac距离大于零模型(p < 2.2×10−16;Kolmogorov-Smirnov检验),表明健康和疾病之间存在显著的聚集。(D)与疾病相关的菌株在肠道中大量存在。箱线图显示了健康人(蓝色)和IBD患者(红色)的微生物群中由健康相关菌株、IBD相关菌株或这两组菌株组合(x轴)所占的百分比(y轴)。箱线图包括:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。
这些菌株进化古老,系统发育多样,且在人类肠道中广泛分布。平均而言,健康与IBD相关菌株在dnaG、gyrB和rpoB位点的同一性差异为1.6%(在16S rDNA中差异为0.15%)。利用分子钟,我们将它们的分化时间定在360万至730万年前(STAR方法),赋予它们数百万年的时间来适应疾病条件。为了确定它们在人类群体中的普遍性,我们使用最大似然法推断了所有样本中每个物种的菌株频率(包括稀有菌株;STAR方法)。令人惊讶的是,疾病富集的菌株被预测在健康肠道中广泛存在,但在疾病中扩展,占健康和疾病状态下所有细菌细胞的20%以上(图2D)。最后,我们将参考基因组整合到菌株系统发育中,以评估系统发育新颖性。尽管健康和疾病相关的菌株属于已知的肠道物种,但它们经常建立以前未观察到的谱系(图2A和S2H)。例如,63%的菌株在RefSeq和UHGG中可用的参考基因组的90%序列同一性范围内没有映射(图S2H)。
为了证明我们的发现对我们的基因集和菌株推断方法具有稳健性,我们使用以下方法重新验证了它们:(1) 来自AMPHORA目录的32个菌株标记基因的更大集合(图S2K、S2L和S4D-S4F);(2) 另一种菌株推断工具StrainPhlAn,它使用从参考基因组中通过计算机模拟得出的特定于分支的标记(图S2I、S2J和S4G-S4I)。最后,尽管我们的统计模型考虑了批次效应,但我们也证明了使用我们最大的队列PRISM中的样本,我们获得了定性上相似的结果(图S2F)。因此,我们的发现对基因集、菌株推断方法和批次效应具有稳健性。
3 疾病关联的系统发育检验不同于基于相对丰度的检验
我们假设,与更传统的差异丰度测试相比,这些系统发育富集测试可能提供一种互补但独特的肠道微生物研究方法。虽然微生物组丰度估计高度可变,并且容易受到批次效应的影响,但单核苷酸多态性(SNPs)是硬编码在细菌基因组中的,因此较少受到技术伪迹的影响,包括DNA提取和PCR扩增中的偏差。这表明系统发育方法可能比差异丰度测试更可靠。
为了更系统地比较系统发育富集和差异丰度测试,我们接下来使用基于相对丰度的混合线性模型,确定了健康和IBD之间的差异丰度物种。该模型控制了年龄、性别、BMI和队列(STAR方法)。我们恢复了许多已知的关联(图S3A),这些关联在队列之间是一致的,因此不是由批次效应的影响(图S3B)。作为一个阴性对照,性别差异与丰度的关联较弱(图S3C)。
在不同物种中,系统发育富集测试和差异丰度测试是不相关的(图3A)。例如,脆弱拟杆菌在健康和IBD中丰度差异显著(p = 1.74×10-6;图3B),但系统发育上并未富集(p > 0.05;图3B)。也就是说,脆弱拟杆菌在疾病中占肠道微生物群的比例更大,但没有菌株与这种增加相关联。相比之下,青春双歧杆菌的不同谱系在健康与IBD中富集(p = 3×10-4;图3D),但它们的相对丰度保持不变(p > 0.05;图3D)。此外,系统发育富集测试捕捉到的分类学谱系与差异丰度测试不同(图3C)。通过利用这些系统发育信号,我们因此可以揭示可能被差异丰度测试忽视的与疾病相关的分支。
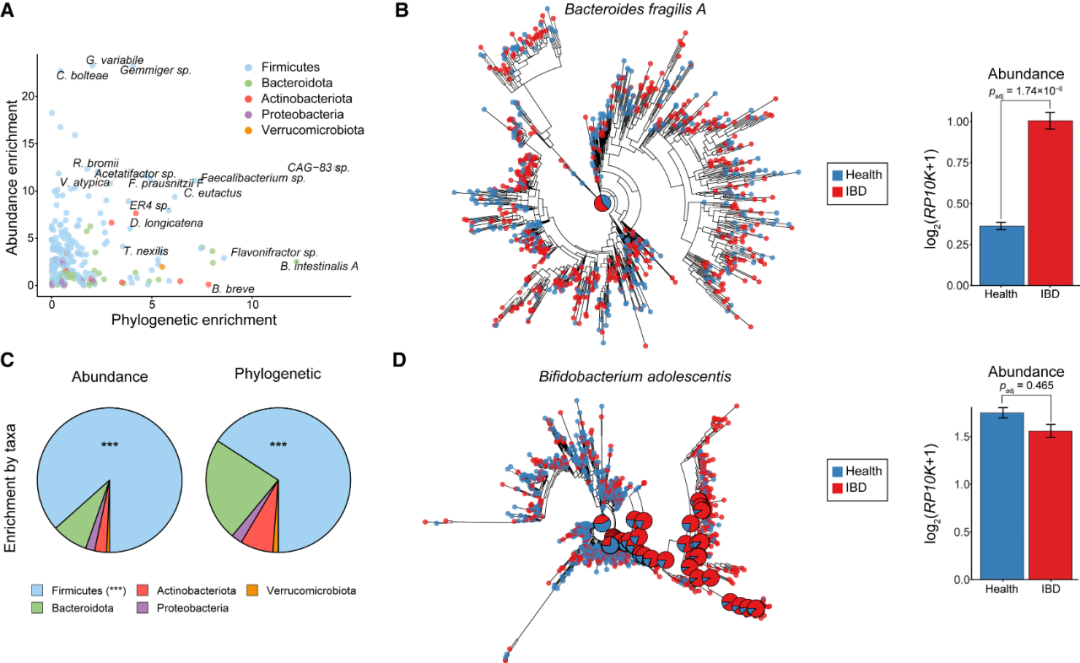 图3. 系统发育富集方法与差异丰度检验不同。(A)所有细菌物种的差异丰度检验( y轴)与系统发育富集检验( x轴)的统计显著性,按门分类着色。这两种测试没有显著相关性(Spearman p= 0.12; p = 0.03)。(B)代表性的例子是脆弱拟杆菌(B. fragilis),其在IBD中丰度差异显著,但在系统发育上没有富集。左侧:脆弱拟杆菌的系统发育在健康或IBD中没有显著富集(校正p值 > 0.05;混合模型)。右侧:脆弱拟杆菌的相对丰度(y轴)在不同疾病状态(x轴)中的变化(校正p= 2×10-6;混合模型)。(C) IBD富集的菌株在系统发育上是不同的。差异丰度(左侧)和系统发育富集测试(右侧)捕获的物种的门分布。差异丰度方法中,厚壁菌门(Firmicutes)所占比例过高(校正p= 6.3×10-16;Fisher检验)。(D)代表性的例子是青春双歧杆菌(B. adolescentis),其在IBD中系统发育上富集,但丰度上没有差异。左侧:青春双歧杆菌的系统发育在疾病中显著富集(校正p = 3×10-4;混合模型)。右侧:青春双歧杆菌的相对丰度(y轴)在不同疾病状态(x轴)中(校正p> 0.05;混合模型)。(B和D) 饼状图:显著节点(校正 p < 0.05;STAR 方法)的疾病状态分布以及所有菌株(根)的背景分布。误差棒:标准误差均值(SEM)
图3. 系统发育富集方法与差异丰度检验不同。(A)所有细菌物种的差异丰度检验( y轴)与系统发育富集检验( x轴)的统计显著性,按门分类着色。这两种测试没有显著相关性(Spearman p= 0.12; p = 0.03)。(B)代表性的例子是脆弱拟杆菌(B. fragilis),其在IBD中丰度差异显著,但在系统发育上没有富集。左侧:脆弱拟杆菌的系统发育在健康或IBD中没有显著富集(校正p值 > 0.05;混合模型)。右侧:脆弱拟杆菌的相对丰度(y轴)在不同疾病状态(x轴)中的变化(校正p= 2×10-6;混合模型)。(C) IBD富集的菌株在系统发育上是不同的。差异丰度(左侧)和系统发育富集测试(右侧)捕获的物种的门分布。差异丰度方法中,厚壁菌门(Firmicutes)所占比例过高(校正p= 6.3×10-16;Fisher检验)。(D)代表性的例子是青春双歧杆菌(B. adolescentis),其在IBD中系统发育上富集,但丰度上没有差异。左侧:青春双歧杆菌的系统发育在疾病中显著富集(校正p = 3×10-4;混合模型)。右侧:青春双歧杆菌的相对丰度(y轴)在不同疾病状态(x轴)中(校正p> 0.05;混合模型)。(B和D) 饼状图:显著节点(校正 p < 0.05;STAR 方法)的疾病状态分布以及所有菌株(根)的背景分布。误差棒:标准误差均值(SEM)
4 与疾病相关的菌株存在于健康人群中,并在慢性炎症期间扩展
在疾病中系统发育丰富的菌株可能是健康肠道的内源性菌株,也可能是从其他人或宿主处获得的外源性菌株。根据进化理论,我们假设内源性菌株将有助于恢复肠道稳态,而外源性菌株则会加剧炎症以获得竞争优势,并增强它们向其他宿主的传播。为了研究菌株的起源,我们假设内源性菌株存在于健康人群中,而外源性菌株不存在。我们首先估计了所有样本中的菌株频率。在这里,“菌株频率”指的是由特定菌株组成的物种的百分比(STAR方法)。健康和IBD相关的菌株在两种健康状态下都被检测到(图2D),这与其潜在的内源性起源一致。
尽管它们似乎有共同的起源,我们假设健康和疾病相关的菌株可能表现出不同的生态动态。例如,共生或病原生物菌株在健康和IBD之间的频率可能是双峰分布,而其他菌株可能保持稳定水平。为了测试这个假设,我们根据菌株在样本中的频率分布进行了聚类,然后确定了与健康、IBD或两者都不相关的菌株簇(图4A;STAR方法)。大多数菌株具有类似对数正态分布,在所有样本中的频率始终较低。然而,健康和疾病相关的菌株具有双峰频率分布,这与共生菌的丧失或病原生物菌的扩张一致,即要么未被检测到,要么极度普遍(图4A;第1组、6组、8组)。其中包括与健康相关的伴生粪球菌(Coprococcus eutactus)菌株和与疾病相关的Tyzzerella nexilis和E. lenta菌株。
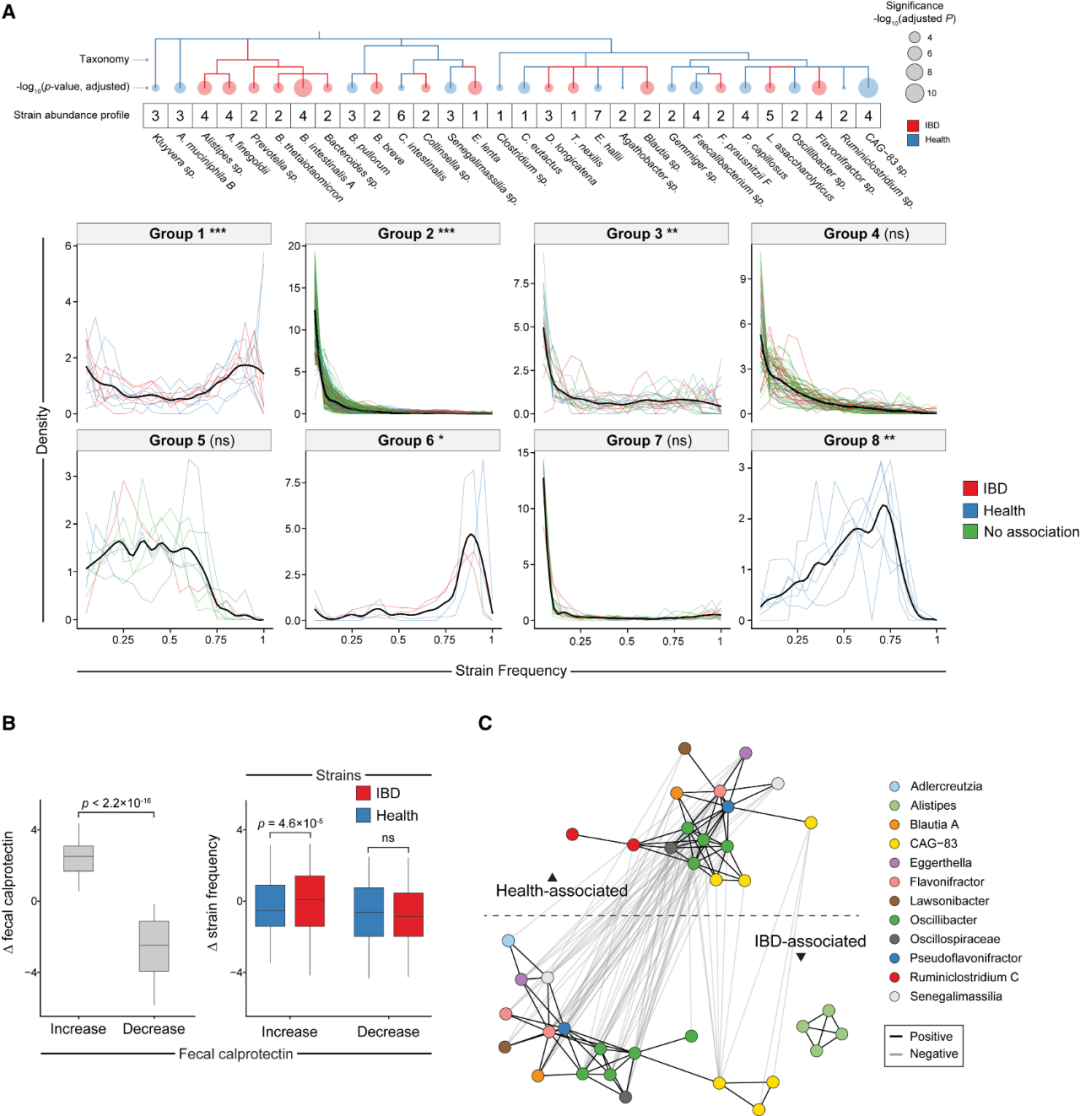 图4. IBD相关菌株在不同患者中具有不同的频率分布和谱系。(A) IBD相关菌株具有不同的频率特征。上图:富集菌株的系统发育(蓝色:与健康相关,红色:与IBD相关,末端:显著性)按丰度特征1-8进行标记。下图:菌株频率特征,展示了所有样本中菌株频率的分布(线条:个别菌株;黑色:平均值)。校正p值:* = 0.05,** = 0.01,*** = 0.001;Fisher检验用于菌株在簇内的富集。基于Wasserstein距离的特征进行聚类。(B) IBD相关菌株在炎症加剧期间具有相对的适应性优势。粪便钙卫蛋白的变化(左侧y轴,log2(x + 1) 转换)以及菌株频率的归一化变化(右侧y轴),作为对粪便钙卫蛋白变化(x轴)的响应。蓝色:与健康相关的菌株;红色:与IBD相关的菌株。箱线图:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。(C)健康和IBD相关菌株在不同患者间共同变化,并形成不同的网络簇。交互网络显示了健康和IBD相关菌株(节点;节点颜色:属)推断频率之间的显著相关性(边;黑色:正相关,灰色:负相关)。虚线:健康菌株与IBD菌株。边:Spearman的相关系数|ρ| > 0.5;校正p< 1×10−8。
图4. IBD相关菌株在不同患者中具有不同的频率分布和谱系。(A) IBD相关菌株具有不同的频率特征。上图:富集菌株的系统发育(蓝色:与健康相关,红色:与IBD相关,末端:显著性)按丰度特征1-8进行标记。下图:菌株频率特征,展示了所有样本中菌株频率的分布(线条:个别菌株;黑色:平均值)。校正p值:* = 0.05,** = 0.01,*** = 0.001;Fisher检验用于菌株在簇内的富集。基于Wasserstein距离的特征进行聚类。(B) IBD相关菌株在炎症加剧期间具有相对的适应性优势。粪便钙卫蛋白的变化(左侧y轴,log2(x + 1) 转换)以及菌株频率的归一化变化(右侧y轴),作为对粪便钙卫蛋白变化(x轴)的响应。蓝色:与健康相关的菌株;红色:与IBD相关的菌株。箱线图:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。(C)健康和IBD相关菌株在不同患者间共同变化,并形成不同的网络簇。交互网络显示了健康和IBD相关菌株(节点;节点颜色:属)推断频率之间的显著相关性(边;黑色:正相关,灰色:负相关)。虚线:健康菌株与IBD菌株。边:Spearman的相关系数|ρ| > 0.5;校正p< 1×10−8。
5 健康和疾病相关的菌株适应炎症环境
尽管这些菌株与人类的疾病状态相关,但它们并不一定适应这些条件。为了更有力地证明它们适应于炎症性疾病,我们假设它们在炎症发作期间会有适应优势,但在炎症的缓解时则没有。然而,菌株适应性的差异在横断面分析中可能难以评估,因为许多因素在患者之间存在差异(例如,宿主遗传学、饮食和微生物-微生物相互作用)。为了解决这个问题,我们在相同的个体内进行了菌株竞争实验,使我们在疾病过程中测量健康与IBD相关菌株的适应性,同时控制病人的变异性。
为了进行这种菌株竞争实验,我们利用了对数百名健康受试者和IBD患者进行了广泛的纵向测量。对于每个受试者和每个物种,我们确定了一对样本,即菌株竞争实验的“初始”和“最终”时间点。我们要求(1)健康和疾病相关的菌株在初始时间点都存在;(2)粪便钙卫蛋白,一种炎症的生物标志物,在时间点之间变化至少50%;(3)时间点间隔超过14天,允许肠道微生物群有足够的时间进行调整。当有多对符合这些标准时,我们选择了粪便钙卫蛋白变化最大的一对。利用这个数据集,我们直接比较了来自66名个体中92多个物种的健康与疾病相关菌株在疾病活动变化(即粪便钙卫蛋白)的适应性。我们测量每个菌株的适应性作为其在初始和最终时间点之间的生长速率。
值得注意的是,健康和疾病相关的菌株都对粪便钙卫蛋白的变化做出了反应。在炎症发作期间,粪便钙卫蛋白水平增加了超过73倍(图4B;p < 2×10-16;Wilcoxon检验),并且与IBD相关的菌株在竞争中胜过了与健康相关的菌株(图4B;p = 5×10-5;Wilcoxon检验)。令人惊讶的是,这种效应在IBD患者中最明显。相反,当炎症活动减退时,粪便钙卫蛋白水平下降了50倍(图4B;p < 2×10-16;Wilcoxon检验),但我们并没有观察到菌株之间的较大差异。这表明与IBD相关的菌株在炎症加剧时具有适应优势,使其能够增加患病率;然而,在缓解期,它们并不恢复到基线;相反,它们会持续数月甚至数年。更普遍地说,这种菌株竞争实验强烈表明这些菌株适应于健康和疾病状态(见讨论)。
最后,鉴于健康和疾病相关的菌株聚集在不同的频率分布中,并对疾病活动的变化做出反应,我们测试了它们是否在IBD患者中共存,它们可能共同产生更大的影响。我们基于样本中菌株频率的相关性构建了一个菌株相互作用网络(考虑了组成性;STAR方法)。健康和IBD相关的菌株形成了高度相关但不同的簇,它们本身是负相关的(图4C)。此外,许多物种的菌株属于健康和IBD富集的簇,它们建立了非常相似的网络拓扑结构。因此,与健康相关的和IBD相关的菌株经常在同一人群中共存,它们本身是负相关的,并且具有一致的网络拓扑结构,这表明了与炎症性疾病相关的不同微生物群“生态型”。
6 与疾病相关菌株的基因组突出了疾病中的重要途径
我们揭示健康与IBD相关菌株之间的遗传差异,以破译其独特的功能。由于区分它们的单核苷酸多态性(SNPs)极少,对近缘菌株进行从头组装是具有挑战性的。然而,对于菌株子集,我们可以通过将参考基因组整合到其系统发育中来推断其基因内容(图S4C;STAR方法)。具体来说,我们根据菌株系统发育中密切相关的参考基因组推断每个菌株的基因内容(STAR方法)。为了解释菌株之间的差异,我们将它们的基因映射到两组途径上:(1) 基因符号前缀,反映操纵子成员身份(例如,“lac”表示乳糖利用基因),以及(2) 从京都基因与基因组百科全书(KEGG)数据库中整理出的自定义途径、模块和酶集(STAR方法)。
对颤螺菌科(Oscillospiraceae) CAG-83 共组装基因组的概念验证应用,确定了核心、健康相关和疾病相关基因的不同集合(图 5A)。有趣的是,菌株差异映射到了炎症疾病生物学的轴线上(图5B)。例如,健康相关菌株编码阿拉伯糖代谢(larBCE)和磷酸酯代谢(phnCDE)的基因,而IBD相关菌株编码异亮氨酸合成(ilvBDK)和铁硫簇组装(sufBCDU)(图5A)(反映了异亮氨酸和铁限制)。合成异亮氨酸(一种支链氨基酸和强效的防御素诱导剂)的能力,表明了免疫调节的潜在途径。扩展到全套KEGG途径,我们发现了必需维生素和细胞表面聚糖合成的额外差异。最值得注意的是,健康相关菌株独特地编码了将叶酸转化为四氢叶酸(THF)的多个步骤,而IBD相关菌株可能将THF转化为其他一碳单元(图5C)。IBD相关菌株在硫胺素和表面聚糖(例如, UDP-糖和dTDP-L -鼠李糖)的生物合成上也存在差异(图5C ),这可能反映了免疫逃避策略。
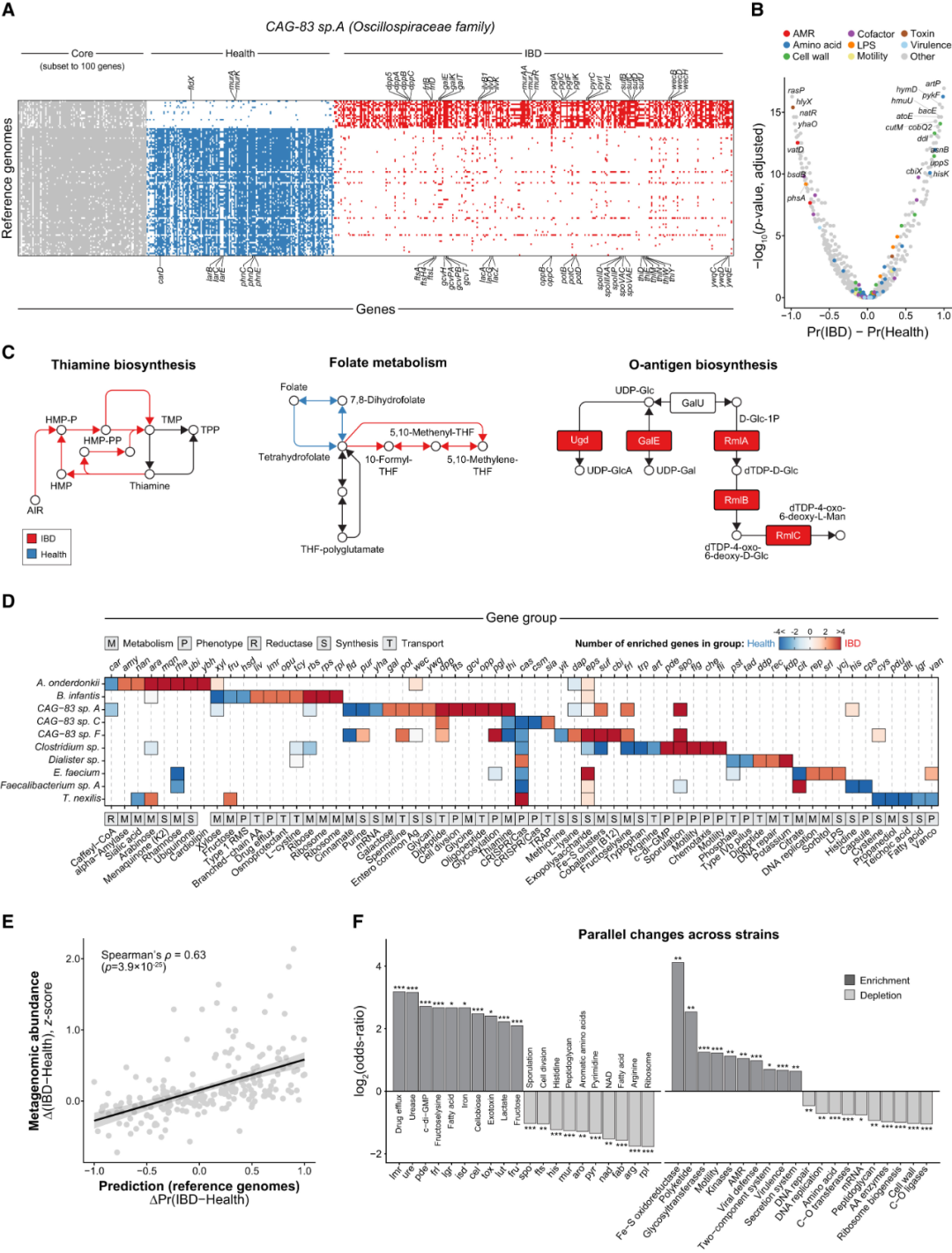
图5. IBD与健康相关菌株之间的遗传差异靶向与疾病相关的途径,并表现出平行的进化变化。(A)对Oscillibacter sp. CAG-83的代表性基因内容分析。热图显示了基因(x轴)在参考基因组(y轴)中的分布。基因被聚类为核心(灰色)、健康相关(蓝色)和疾病相关(红色)组。选择提供的基因注释。(B) IBD与健康相关CAG-83菌株的基因内容。火山图显示疾病富集(x轴)和统计显著性(y轴)(STAR 方法),带有功能注释(颜色)和选定的基因标记。(C) CAG-83健康与IBD相关菌株的生物合成差异。途径图显示了在健康(蓝色)与IBD相关菌株(红色)之间不同的酶,这些酶针对硫胺素、叶酸和O-抗原多糖生物合成的几个步骤。(D) IBD相关菌株中富集的操纵子。对于每个基因符号前缀(列;STAR 方法),在细菌物种(行)中显著富集的基因数量在IBD(红色)与健康相关(蓝色)菌株之间。为每个操纵子(列;底部)提供了功能注释。(E) 宏基因组验证基因内容差异。对于在健康或IBD相关菌株中显著富集的操纵子(灰色点),在与健康和IBD菌株相关的宏基因组样本中操纵子内基因的相对丰度的平均差异(y轴),与仅从参考基因组预测的差异(x轴)进行比较。相对丰度是针对每个样本中的核心基因进行归一化,然后在样本间进行标准化(STAR 方法)。黑色线,最佳线性拟合。(F) 菌株的遗传差异在与疾病相关的途径中显示出并行的进化变化。对于在健康或IBD相关菌株中富集的操纵子(左侧)和途径(右侧)(x轴),疾病富集的几率比(y轴)。颜色:深灰色:富集;浅灰色:减少。校正p值:* = 0.05,** = 0.01,*** = 0.001(Fisher检验;STAR 方法)。为每个操纵子提供了功能注释。
接下来,我们预测了 59 种健康相关菌株和 IBD 相关菌株的基因组(图5D)。尽管IBD相关参考基因组序列的可用性有限,但我们仍然将菌株的遗传差异映射到许多与疾病相关的途径上(图5D)。具体来说,我们发现了辅酶Q合成(mqn操纵子;昂氏别样杆菌(Alistipes onderdonkii))、鞭毛(flg操纵子;梭菌属(Clostridium sp.))和万古霉素抗性(van操纵子;T. nexilis)的变化。综合所有59个物种,我们发现在运动途径(例如,T. nexilis和Clostridium sp.中的鞭毛)、致病性(例如,Clostridium和普雷沃氏菌属(Prevotella sp.)中的溶血素)以及脲酶(例如,布劳特氏菌属(Blautia)和另枝菌属(Alistipes sp.)中的脲酶操纵子)发生了平行的进化变化(图5F和S5B;STAR方法)。健康和疾病相关菌株之间通常存在差异的其他途径包括抗生素抗性(lmr操纵子)、从血红素中铁清除(isd操纵子)以及氧化还原酶。作为阴性对照,管家基因的功能在菌株中显著减少,包括DNA复制、DNA修复和核糖体生物合成。最后,我们从FoldSeek中鉴定了结构同源蛋白簇的平行变化,包括Ton B外膜蛋白和柠檬酸铁调节的反复基因组变化(图S5B),从而揭示了可能导致炎症性疾病的假定适应性。
为了验证我们对菌株基因组的预测,我们测试了是否可以仅使用我们的基因组预测来准确估计宏基因组中基因的相对丰度(图5E)。重要的是,这些预测不依赖于对宏基因组基因内容的任何知识。值得注意的是,我们对健康与IBD相关菌株之间基因内容差异的预测对其宏基因组丰度具有很强的预测性(Spearman相关系数 r = 0.63; p < 2×10-16)。例如,我们预测与疾病相关的Clostridium sp.菌株编码了fli操纵子(图5D);一致地,fli基因在与IBD相关的宏基因组中富集(图5E)。因此,与疾病相关的菌株可能已经获得了一系列在炎症中持续存在的策略,包括抗氧化应激、免疫逃避和致病性;重要的是,这些变化不一定代表它们在疾病中的适应性决定因素。
7 健康和疾病相关的菌株与疾病严重程度的生物标志物有关
为了评估这些菌株是否在疾病的诊断或治疗中发挥作用,我们测试了它们是否可以用于预测疾病状态、疾病亚型或疾病严重程度。疾病严重程度通过粪便钙卫蛋白的水平进行评估,粪便钙卫蛋白是粘膜炎症的生物标志物,与肠道炎症严重程度的临床评估密切相关。我们训练模型根据不同细菌组的相对丰度预测这些特征:健康相关菌株(n = 66);IBD相关菌株(n = 66);非相关菌株(n = 66);所有菌株(n = 198);有菌株估计的物种(n = 66);以及所有物种(n = 535)(STAR方法)。我们专注于PRISM,这是唯一一个具有足够的菌株和粪便钙卫蛋白测量值的队列。重要的是,这些模型专注于单一队列,并且基于相同的输入训练集(即66个具有菌株估计的物种),从而考虑了批次效应和模型复杂性。
对于疾病状态和亚型的预测,物种模型优于菌株模型(图6A、6B、S6A和S6D)。然而,健康相关菌株在预测IBD严重性方面表现出色(图6A和S6A),其预测与粪便钙卫蛋白水平相关(图6C)。这一发现对模型选择(随机森林、梯度提升回归;图S6A)和模型参数(图S6B)具有稳健性。为了确定推动这一预测的细菌菌株,我们评估了表现最佳的模型中每个菌株的“特征重要性”(图6D),并确定了比其相应物种更重要的菌株(图6E)。与健康相关的E. lenta菌株(图S4B)是菌株模型中最显著的特征,但在物种模型中不是(图6D和6E),并且与严重性呈负相关(图S6C)。实际上,健康相关E. lenta菌株的相对丰度,而不是其他菌株或物种,与所有样本中的炎症严重程度呈负相关(图6F)。为了在PRISM之外的其他队列中测试这种关联,我们确认E. lenta菌株在许多队列中在健康受试者中富集,在IBD患者中减少(图S6E)。
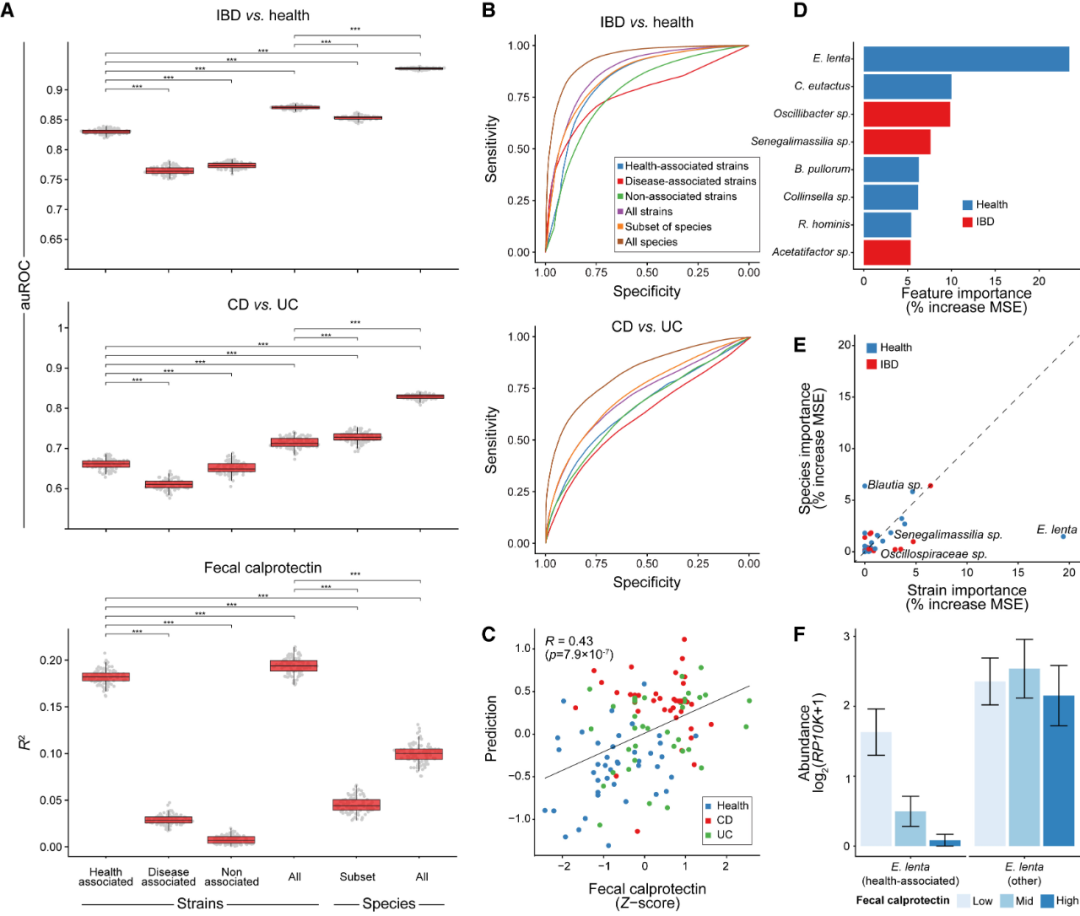
图6. 健康和IBD相关菌株能准确预测炎症的生物标志物。(A和B) 菌株模型在预测疾病严重性方面优于物种模型。(A) 箱线图显示了不同菌株或物种的随机森林模型(x轴)在预测疾病状态(顶部)、疾病亚型(中间)和疾病严重性(底部)时,接收者操作特征(ROC)曲线下面积或解释的方差(R2;y轴)。箱线图:25%、50%和75%分位数;须线:1.5倍四分位间距(IQR)。*p = 0.05,**p = 0.01,***p = 0.001;Wilcoxon检验。(B) ROC曲线展示了不同菌株和物种的随机森林模型(颜色)在预测疾病状态(顶部)和疾病亚型(底部)时的灵敏度(y轴)与特异性(x轴),在不同的分类阈值截断点下。(C)与健康相关的菌株能准确预测疾病严重性。不同疾病状态(颜色)的PRISM队列样本(圆点)的粪便钙卫蛋白预测水平( y轴)与实测水平( x轴)的散点图。黑色线,最佳线性拟合。(D) 菌株(y轴)在预测疾病严重性方面的重要性(x轴),通过从模型中移除菌株后均方误差的增加来衡量。结果展示了“所有菌株”模型。颜色:蓝色,与健康相关的菌株;红色,与IBD相关的菌株。(E) E. lenta菌株的丰度在预测疾病严重性方面比物种丰度更重要。物种的重要性(y轴)与菌株的重要性(x轴)对比,按疾病关联的方向着色(红色:IBD,蓝色:健康)。(F)与健康相关的E. lenta菌株与疾病严重性相关。基于粪便钙卫蛋白的三等分法,在不同粪便钙卫蛋白水平的样本中,健康相关和所有其他E. lenta菌株的平均丰度(y轴)(x轴)。误差条:标准误差均值(SEM)。
为了推断E. lenta的基因内容,我们试验了一种基于共丰度基因组的方法。尽管这种方法识别 E. lenta 基因的灵敏度超过 94%,但它也缺乏精确度;因此,精确定位的基因是初步的,需要实验验证(图S5C;STAR方法)。尽管如此,我们将68,503个基因映射到E. lenta菌株上(表S6)。最重要的发现揭示了健康相关E. lenta菌株与胆汁盐水解酶的潜在关联,以及与疾病相关菌株和抗氧化剂(如过氧化氢酶和生育酚环化酶)的关联。这些结果表明在胆汁酸代谢中的关键作用以及对氧化应激的适应,并与E. lenta在过氧化氢酶和胆汁盐降解活性异质性的研究一致。综上所述,我们的研究表明,E. lenta与 IBD的先前关联很可能是菌株特异性的,并且 E. lenta 菌株可以作为生物标志物,甚至可以预防疾病。
讨论
通过利用微生物组中的进化信号,我们的工作揭示了炎症性疾病中一个隐藏的与疾病相关的菌株库(图1),这在过去的研究中未被发现(图3)。我们发现了数百种与健康和 IBD 相关且可能适应健康和 IBD 的不同菌株(图2)。与过去专注于丰度差异细菌的研究不同,我们的方法确定了与疾病具有长期进化关联的细菌谱系。总的来说,这些菌株占肠道细菌的很大一部分(图2)。我们的分子钟估计这些菌株在数百万年前就已分化,这可能与原始人类的出现有关。然而,分子钟是嘈杂和难以校准的,这些菌株在哺乳动物分化过程中可能更晚或甚至更早发生了分化。
通过在疾病过程中追踪同一个体内部的菌株竞争,我们展示了与疾病相关的菌株在炎症加剧期间具有相对的适应性优势(图4),从而暗示了其对疾病的适应性。实际上,健康和IBD相关菌株比其各自的物种更能强烈预测炎症的强度(图6)。特别是,与健康相关的E. lenta菌株与疾病活动呈负相关(图6),这表明它们可以作为生物标志物,或者可能在疾病中发挥保护作用。
这些菌株可以适应与IBD相关的各种情况,包括免疫反应、代谢变化、药物摄入、饮食,甚至对疾病的遗传倾向。事实上,通过重建健康和疾病相关菌株之间的遗传差异,我们展示了它们在生态学上的多样性,并在包括氧化应激、营养生物合成、抗生素抗性以及细胞壁在内的多种途径中发生了平行变化(图5)。这表明与疾病相关的一系列条件,而不是任何单一的选择力量(如抗生素),选择了适应这些条件的不同菌株。这些遗传差异的一个子集甚至可能影响宿主,包括推定的毒力因子(如粘附素和溶血素),这可能有助于细菌的持续存在,甚至加剧IBD。通过揭示这些遗传差异,我们为细菌在炎症期间可能采用的潜在分子策略提供了见解。未来的实验工作测试这些预测可能会发现新的微生物-宿主相互作用,从而塑造疾病风险。
与疾病相关的菌株也在健康受试者中被发现,但它们并不反映主导菌株(图2)。这些菌株可能源自健康肠道的内源性储库(图4),例如结肠隐窝、阑尾、回肠、淋巴组织斑块,甚至远端身体部位(例如口腔)。它们还可能提供定植抗性,即IBD相关的共生菌株占据炎症生态位,以防止其他细菌入侵者的机遇性定植。值得注意的是,其他菌株仅在IBD患者中发现,而在健康对照组中很少见,这可能是由于疾病开辟了新的生态位。这些发现突出了形成和主导这些菌株出现的多种生态驱动因素,并强调了对IBD相关菌株进行功能剖析的迫切需求(图4和5)。
在健康受试者中也发现了与疾病相关的菌株,但并未体现出优势菌株(图2 )。这些菌株可能来源于健康肠道中的内源性储存库(图4 ),如结肠隐窝、盲肠、回肠、淋巴小结,甚至远端身体部位(如口腔)。它们还可以提供定植抗性,使IBD相关的共生菌菌株占据炎症生态位,以防止其他细菌入侵者的机会性定植。值得注意的是,其他菌株仅在IBD患者中发现,而在健康对照中很少发现,这可能是由于疾病开辟了新的生态位。这些发现强调了影响这些菌株出现和优势地位的各种生态驱动因素,并强调了对IBD相关菌株进行功能解剖的迫切需求(图4和图5)。
总的来说,我们定义了一种利用肠道微生物组的进化信号来发现可能适应健康和疾病的菌株的方法,以解码它们的基因组适应性,并将它们与临床表型联系起来。我们的工作为系统解剖肠道微生物提供了路线图,可以扩展到确定其他特征的微生物基础,如类风湿性关节炎、I型糖尿病和癌症免疫治疗的反应,并最终揭示驱动或维持复杂免疫介导疾病的机制。
原文链接:https://doi.org/10.1016/j.chom.2024.05.022


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









