






点击蓝字 关注我们
多组学揭示肥胖易感与肥胖抵抗小鼠的关键特征

iMetaOmics主页:http://www.imeta.science/imetaomics/
研究论文
● 原文链接DOI: https://doi.org/10.1002/imo2.59
● 2025年2月4日,浙江大学朱善宽、杨菲等在iMetaomics在线发表了题为“Multi-omics reveals different signatures of obesity-prone and obesity-resistant mice”的文章。
● 本研究整合微生物组、代谢物和转录组数据,系统解析了宿主基因、代谢及肠道微生物组在肥胖易感(OP)和肥胖抵抗(OR)中的作用,揭示了关键菌属及其与胆汁酸代谢、氨基酸谱和肠道屏障功能的互作关系,为理解肥胖易感和肥胖抵抗的表型差异机制提供了重要参考。
● 第一作者:王聪聪、林锦华、段萌
● 通讯作者:朱善宽(zsk@zju.edu.cn)、杨菲(yangfei919@zju.edu.cn)
● 合作作者:何佳玲、哈丽热则·斯马义、陈宁馨、陈心愉、焦靥、何威、Kenneth A Dyar
● 主要单位:浙江大学医学院公共卫生学院、浙江大学医学院附属儿童医院、国家儿童健康临床医学研究中心、浙江大学慢性病研究所、浙江大学滨江研究院、德国亥姆霍兹慕尼黑研究中心、德国糖尿病研究中心 (DZD)、慕尼黑工业大学医学与健康学院
亮 点
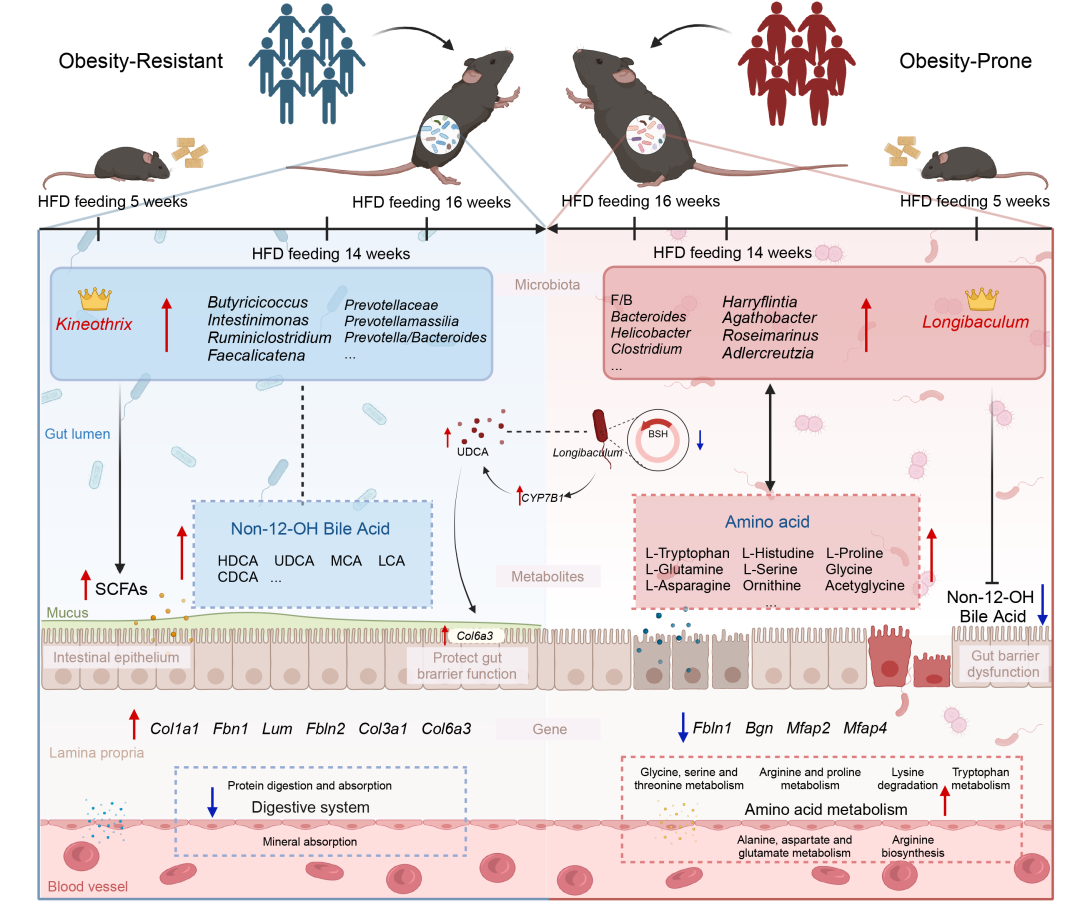
● 描述了肥胖易感(OP)和肥胖抵抗(OR)小鼠的肠道微生物群动态变化,并鉴定出Longibaculum和Kineothrix为关键菌属(keystone bacteria);
● Kineothrix、Intestinimonas和Fournierella在OR小鼠中显著富集,同时也在肥胖抵抗人群中富集;
● 10 种非12-羟基胆汁酸(包括熊去氧胆酸和猪去氧胆酸)在OR小鼠中显著升高,并能减少高脂饮食诱导的体重增长;
● 发现 22 种特定氨基酸谱 作为肥胖易感性的潜在生物标志物。
摘 要
肥胖易感(Obesity-prone, OP)和肥胖抵抗(Obesity-resistant, OR)个体表现出显著的代谢差异,这可能受到肠道微生物组变异的影响。然而,宿主-微生物组相互作用对肥胖易感性的影响仍不明确。本研究采用整合多组学方法,探索高脂饮食(HFD)喂养的 OP 和 OR 小鼠在微生物、代谢和遗传方面的差异,并分析了OP 和 OR人群肠道微生物组的变化情况。在 OP 小鼠中,动态变化的肠道微生物群以 Longibaculum 稳定存在为特征,而 OR 小鼠则以 Kineothrix 为优势菌属,我们将二者定义为关键菌。此外,八种优势菌属与胆汁酸代谢物和氨基酸显著相关。其中,有三种菌属在人类 OR 组中也被鉴定,并与可能支持肠道屏障功能的基因呈正相关。我们进一步鉴定了 22 种特定氨基酸谱 作为肥胖易感性的潜在生物标志物,同时发现 OR 小鼠粪便中 10 种非 12-羟基胆汁酸 含量显著增加。体内小鼠实验表明,熊去氧胆酸(ursodeoxycholic acid)和猪去氧胆酸(hyodeoxycholic acid)可降低 HFD 诱导的肥胖。此外,OP 小鼠的结肠中炎性细胞含量较高。这些研究结果表明,宿主-微生物组相互作用可能导致 OP 和 OR 表型的差异。本研究提供了与肥胖相关的重要肠道标志物,为深入理解肥胖易感和抵抗表型提供了重要参考。
视频解读
Bilibili:https://www.bilibili.com/video/BV12nPheeETg/
Youtube:https://youtu.be/6uc5yZoq6CI
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/imetaomics/
全文解读
引 言
肥胖是全球范围内广泛存在的健康问题,也是2 型糖尿病、心血管疾病及某些癌症的重要风险因素。除了遗传调控外,环境因素,尤其是高热量饮食的过度摄入,在肥胖的发展过程中起着至关重要的作用。Chang 等研究发现,即使接受相同的高脂饮食(HFD),大鼠的体重表型仍存在显著差异。其中,一些大鼠更容易出现显著体重增加(肥胖易感,Obesity-Prone, OP),而另一些则对肥胖具有抗性(Obesity-Resistant, OR)。类似的现象也在小鼠中得到了验证。目前,OP 和 OR 个体之间的差异性仍未被充分阐明,但该问题对肥胖研究具有重要意义,且随着多组学(-omics)方法的发展,该领域研究取得了显著进展。在 OP 和 OR 小鼠中,肠道微生物群特征存在显著差异,例如 Oscillibacter 和 Clostridium 在 OP 小鼠中的丰度不同于 OR 小鼠。此外,将 OP 小鼠的微生物群移植至无菌小鼠体内,可诱导其表现出 OP 小鼠的表型特征,这进一步凸显了探索关键菌属在 OP 和 OR 形成中的作用的重要性。
此外,多组学数据整合已被用于探索 OP 和 OR 相关机制。研究表明,肠道菌群-胆汁酸轴的失调可能促进肥胖的发生;在 OP 大鼠中,代谢通路的改变可能影响脂质代谢;同时,“微生物-肠道-大脑”轴可能在肥胖抵抗中发挥重要作用。肠道菌群可能通过调节宿主的生理适应性来影响其表型。具体而言,肠道微生物能够通过其代谢产物(如胆汁酸、短链脂肪酸、氨及其他生物活性化合物)广泛影响机体的代谢系统。这些代谢物可被肠肝循环吸收进入血液循环并参与细胞信号传导和增殖,进而影响生理功能。这些过程可能导致肠道黏膜结构、屏障完整性及免疫活性的改变,最终塑造宿主的代谢表型。
本研究旨在利用整合多组学方法(结合微生物组学、代谢组学和转录组学),探索宿主-微生物互作与 OP 和 OR 小鼠表型差异之间的关系,探索肠道菌群的动态变化及其在肥胖易感或抵抗中的潜在机制。
结 果
OP 和 OR 小鼠的表型差异
不同阶段小鼠的体重变化如 图 1A 所示。经过 3 周高脂饮食(HFD)喂养后,OP 小鼠的体重增长显著高于 OR 小鼠(8 周龄,p < 0.05)。随着喂养时间的延长,这一差异更加明显。16 周(21 周龄)时,对小鼠肝脏和脂肪组织进行 苏木精-伊红染色后发现,其组织结构和细胞形态发生了明显变化(图 1B)。OP 小鼠的肝细胞明显水肿,多数细胞表现出中度至重度的弥漫性脂肪变性,主要表现为大泡性和小泡性脂肪变性。此外,仅观察到少量淋巴细胞浸润。相比之下,OR 小鼠的肝细胞胞浆呈颗粒状,部分细胞出现小空泡,但其数量显著少于 OP 小鼠(p < 0.001),且多于正常肝脏(图 S1,表 S1)。白色脂肪组织变化:OR 小鼠白色脂肪组织中的脂滴显著小于 OP 小鼠(p < 0.001),但仍大于对照组(Con 组)(图 S1,表 S1)。
OP 和 OR 小鼠肠道微生物群的动态变化
在 0、2 和 5 周的高脂饮食(HFD)喂养过程中,OP 和 OR 小鼠的肠道微生物群 α- 和 β-多样性未表现出显著差异。然而,在 14 和 16 周的 HFD 喂养后,两组小鼠的肠道菌群多样性出现了显著差异(p < 0.05)(图 1C、D)。使用 线性判别分析效应量(LEfSe)方法对 OP 和 OR 小鼠的关键菌属 进行了鉴定(图 1E)。0 和 2 周 HFD喂养小鼠,未观察到两组小鼠在菌属水平上的显著差异。经过5 周 HFD喂养小鼠,在 OP 和 OR 小鼠中分别出现了 Longibaculum 和 Kineothrix 两个显著不同的菌属(p < 0.05),并被认为是 关键菌属(keystone bacteria)。这是OP 和 OR 小鼠首次表现出差异性菌属。14 和 16 周 HFD喂养,OP 小鼠中优势菌属:Longibaculum、Agathobacter、Roseimarinus、Harryflintia 和 Adlercreutzia 稳定且重复出现。OR 小鼠中优势菌属:Kineothrix、Butyricicoccus、Intestinimonas、Ruminiclostridium 和 Faecalicatena 稳定且重复出现(图 1E)。此外,我们使用 qPCR 对这些结果进行了验证,证实了菌属差异的显著性(图 S2,表 S2)。14 和 16 周的 HFD 喂养阶段 被选定为 OP 和 OR 小鼠表型在长期饮食干预后趋于稳定的阶段。
人群研究中的肠道微生物群
在 OP 和 OR 人群组之间观察到显著的肠道微生物群 β-多样性差异(p < 0.05,图 1F)。两组人群的基线特征详见 表 S3。此外,在 OR 小鼠中富集的 Kineothrix、Intestinimonas 和 Fournierella 三种菌属同样在 OR 人群中被检测到(图 1G)。

图1. 肥胖易感(OP)和肥胖抵抗(OR)小鼠及人群的肠道微生物群变化
(A) 不同时间点的体重变化。数据采用双因素方差分析(ANOVA) 进行统计分析(因素:组别和时间)。组间显著性差异标注为 (*p < 0.05, **p < 0.01, *p < 0.001),OP 组 vs. OR 组,OP 和 OR 组 n = 10,对照组(Con)n = 5。(B) 附睾白色脂肪组织(eWAT)和肝组织的苏木精-伊红(H&E)染色代表性图像,箭头指示脂肪小泡或炎性细胞的存在。(C) 不同时间点 OP 和 OR 小鼠粪便微生物群的 Shannon 指数和 Simpson 指数(n = 10/组),时间点:0、2、5、14 和 16 周 HFD。统计分析采用 Mann-Whitney U 检验,*p < 0.05。(D) OP 和 OR 小鼠不同时间点粪便微生物群组成的主坐标分析(PCoA,n = 10/组),时间点:0、2、5、14 和 16 周 HFD。(E) OP 和 OR 小鼠高脂饮食(HFD)喂养 5、14 和 16 周后微生物丰度的差异性分析(LEfSe)。线性判别分析(LDA)效应值显示了在 OP 和 OR 组微生物群中丰度显著差异的优势菌属(n = 10/组),LDA 评分 > 2。(F) OP 和 OR 人群的 Bray-Curtis 距离主坐标分析(PCoA,p = 0.001)。Permanova 分析用于调整混杂因素,包括性别、年龄、吸烟状况、饮酒、教育水平和体脂率。(G) LEfSe 分析 OP 和 OR 人群中丰度存在显著差异的微生物。图中展示了具有统计学显著性差异的菌属(FDR < 0.1,LDA 评分 > 2)。
OP 和 OR 小鼠血清代谢组的差异
在 OP 和 OR 小鼠 中,共鉴定出 1567 种血清代谢物。正交偏最小二乘判别分析(OPLS-DA) 模型成功区分了两组小鼠的代谢谱特征(图 2A)。结合变量投影重要性(VIP)、倍数变化(fold change)和单变量分析的 q 值,共鉴定出 79 种差异代谢物,其中 42 种在 OP 小鼠中水平升高,37 种在 OR 小鼠中水平升高(图 2B,表 S4)。与 OR 小鼠相比,OP 小鼠中升高的代谢物主要包括:氨基酸及其相关代谢物、有机酸、吡啶及其衍生物。而 OP 小鼠中降低的代谢物主要包括:胆汁酸、醇类及其衍生物、黄酮类化合物(表 S5)。图 2C 显示了差异代谢物的聚类分析,聚类热图直观展示了两组小鼠差异代谢物的表达水平。使用 京都基因与基因组百科全书(KEGG)数据库 进行通路分析,发现差异代谢通路主要涉及:氨基酸及其他氨基酸代谢、辅因子与维生素代谢、神经系统、能量代谢、消化系统、碳水化合物代谢(图 2D,表 S6)。与 OR 小鼠相比,OP 小鼠中 17 条代谢通路的整体表达水平升高,3 条代谢通路的表达水平降低(图 2E)。其中,氨基酸代谢占比最高,包括:β-丙氨酸代谢、D-谷氨酰胺和 D-谷氨酸代谢、精氨酸生物合成、丙氨酸、天冬氨酸和谷氨酸代谢(p < 0.01)。
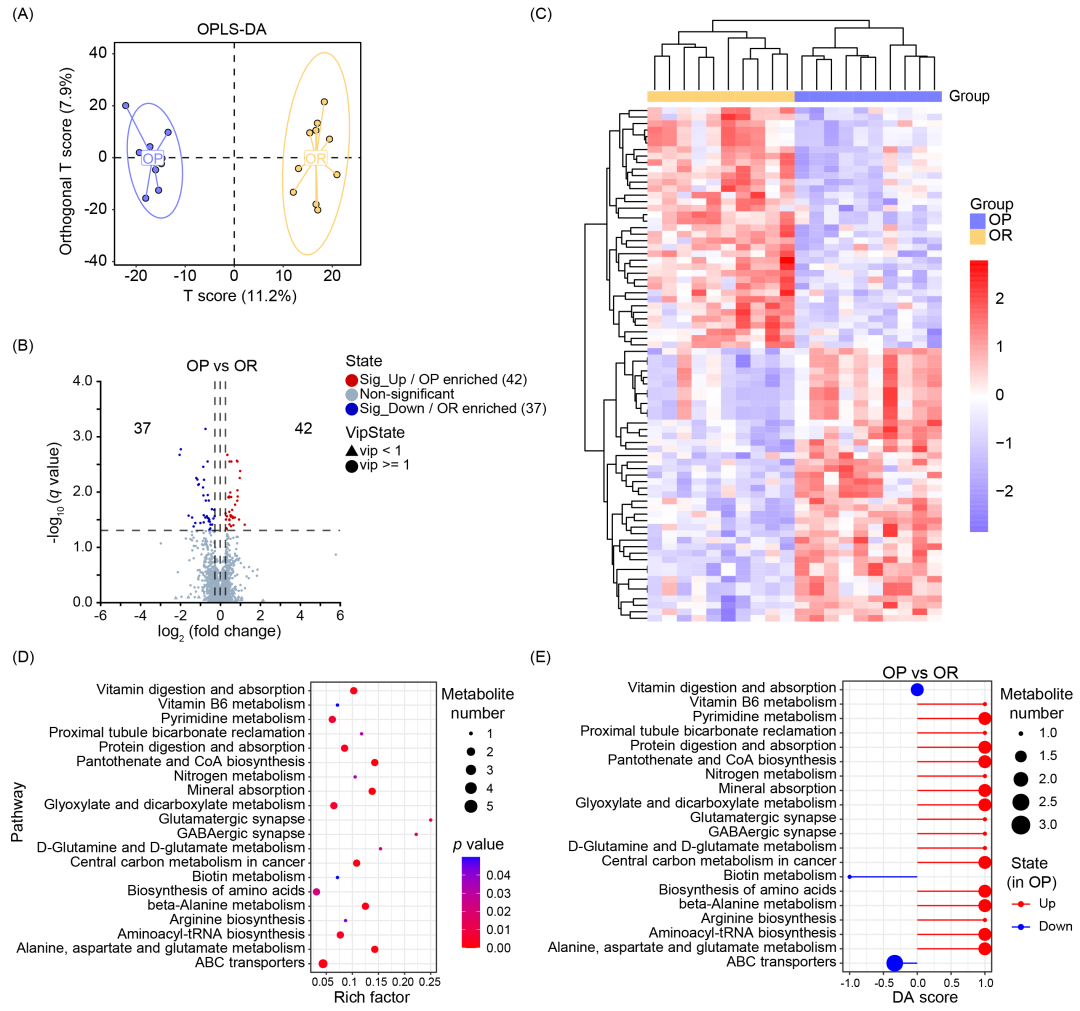
图2. OP 和 OR 小鼠的血清代谢组变化
(A) 正交偏最小二乘判别分析(OPLS-DA) 结果,展示 OP 和 OR 小鼠血清代谢物组成的差异(n = 10/组)。(B) 火山图(Volcano plot) 展示 OP 和 OR 小鼠血清代谢物的差异表达情况。蓝色表示显著下调的代谢物,红色表示显著上调的代谢物,灰色表示无显著差异的代谢物(n = 10/组)。(C) 差异代谢物的表达热图。基于 OP 和 OR 小鼠的差异代谢物表达水平 进行聚类分析。(D) 代谢通路富集分析的气泡图。(E) 20 条 KEGG 代谢通路的差异丰度评分(DA 评分)。红色表示该通路中的所有差异代谢物在 OP 组中表达上调。蓝色表示该通路中的所有差异代谢物在 OP 组中表达下调。圆点大小表示该通路中代谢物的数量,圆点越大,说明该通路中代谢物数量越多。
OP 和 OR 小鼠粪便代谢组的差异
本研究对粪便样本进行了代谢组学分析,涵盖了氨基酸、有机酸、脂肪酸和糖类。此外,还定量分析了 400 多种小分子代谢物,包括 胆汁酸、肉碱、苯基和苄基衍生物,以及吲哚化合物。OPLS-DA 模型 成功区分了 OP 和 OR 小鼠的粪便代谢物谱(图 3A)。共鉴定出 62 种显著差异的代谢物(图 3B)。其中 22 种属于氨基酸类,包括 L-色氨酸(L-tryptophan)和 N-乙酰丝氨酸(N-acetylserine)(图 3C,S3)。所有这些氨基酸在 OP 小鼠中显著升高。这些代谢物在预测 OP 代谢表型方面表现良好(曲线下面积 AUC > 0.75,图 S3)。在 62 种差异代谢物 中,有 10 种属于胆汁酸类,包括 β-熊去氧胆酸(β-UDCA)和 β-猪去氧胆酸(β-HDCA)(图 3D)。所有胆汁酸在 OR 小鼠中显著升高。喂养 HFD + HDCA 或 UDCA 8 周的小鼠 相较于仅喂 HFD 组,体重增长显著减少,并伴随结肠 mRNA(Col6a3 和 Cyp7b1)表达水平显著上调(图 3E, F)。另外,Col6a3 和 Cyp7b1 也在 OR 小鼠的结肠转录组中高表达(表 S7,图 S4)。代谢物相关性分析(图 S5)显示显著差异的氨基酸(红色标记)和胆汁酸(蓝色标记)呈负相关。KEGG 代谢通路富集分析(图 3G,表 S8)得到,差异代谢物主要涉及:氨基酸代谢、碳水化合物代谢、消化系统、能量代谢、神经系统。KEGG 代谢通路整体变化分析(图 3H)显示,与 OR 小鼠相比,OP 小鼠中 20 条代谢通路显著上调。
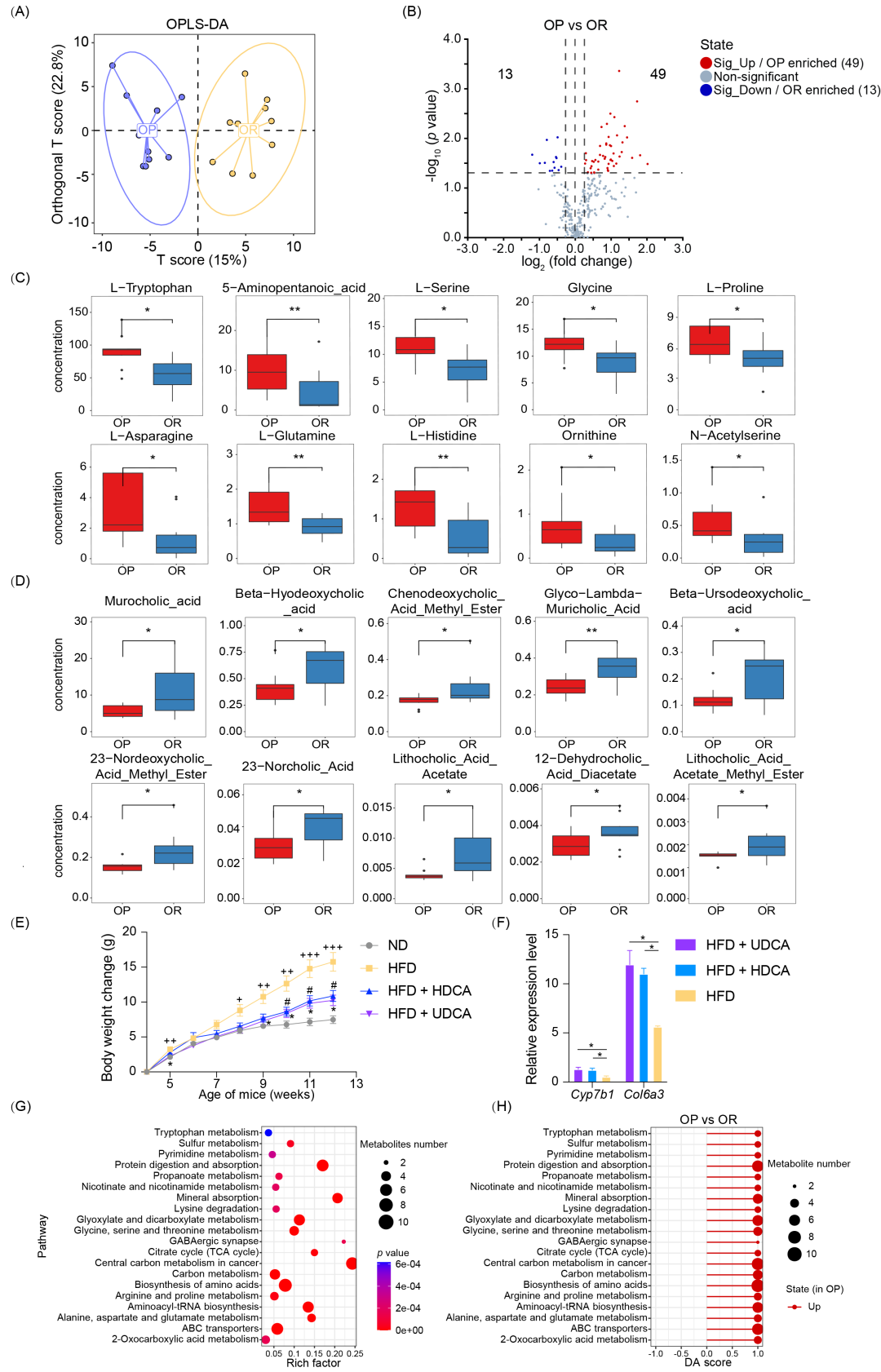
图3. OP 和 OR 小鼠粪便代谢组的变化
(A) 正交偏最小二乘判别分析(OPLS-DA) 展示 OP 和 OR 小鼠的粪便代谢物组成差异(n = 10/组)。(B) 火山图(Volcano plot)展示 OP 和 OR 小鼠粪便代谢物的差异表达情况。蓝色:显著下调的代谢物。红色:显著上调的代谢物。灰色:无显著差异的代谢物(n = 10/组)。(C, D) 差异代谢物的箱线图。(C) 氨基酸(p < 0.05)(D) 胆汁酸(p < 0.05)。Welch’s t 检验(n = 10/组)。(E) 高脂饮食(HFD)+ 熊去氧胆酸(UDCA)或猪去氧胆酸(HDCA)对小鼠体重变化的影响。采用双因素方差分析(ANOVA) 进行统计分析(因素:组别和时间)。+p < 0.05;++p < 0.01;+++p < 0.001(HFD 组 vs ND 组)。#p < 0.05(HFD + HDCA 组 vs HFD 组)。*p < 0.05;**p < 0.01(HFD + UDCA 组 vs HFD 组)(F) 体内实验:结肠 mRNA(Col6a3, Cy7b1)表达水平的 qRT-PCR 验证。非配对 Student’s t 检验(n = 3),*p < 0.05。(G) KEGG 代谢通路富集分析的气泡图(基于粪便代谢物)。气泡大小表示通路中注释到的差异代谢物数量,气泡越大,说明该通路中差异代谢物越多。(H) KEGG 代谢通路差异丰度评分(DA 评分)(OP vs OR)。红色表示 OP 组中该通路的所有差异代谢物表达上调。圆点大小表示该通路中代谢物的数量,圆点越大,说明该通路中代谢物数量越多。
OP 和 OR 小鼠结肠转录组的差异
为探讨结肠核心菌群对宿主的干扰或保护机制,本研究对 OP 和 OR 小鼠的结肠组织 进行了 RNA 测序,以量化基因表达特征。共检测到 17,778 个基因(图 4A)。采用 Gene Ontology(GO)富集分析 对差异表达基因(DEGs)进行功能注释(判定标准:fold change ≥ 1.2 或 ≤ 0.83,q-value < 0.2)。在所有差异表达基因中,相比 OR 小鼠,10 个 mRNA 在 OP 小鼠中上调,101 个 mRNA 在 OP 小鼠中下调(图 4B)。GO 功能分析显示,OP 小鼠的富集通路主要涉及免疫反应和肠道结构损伤,包括:内皮屏障形成的负调控、黏附连接组织的负调控、钙粘蛋白介导的细胞-细胞粘附负调控、细胞-细胞粘附负调控(图 4C)。OR 小鼠的富集通路主要涉及细胞粘附和极性维持,包括:细胞粘附、细胞极性建立或维持的调控、肾小球系膜细胞-基质粘附、细胞-基质粘附、细胞-细胞粘附、细胞-基质粘附的正向调控(图 4D)。差异基因筛选及关键驱动基因(KDA)分析,采用更严格标准(fold change ≥ 1.2 或 ≤ 0.83,q-value < 0.05),共筛选出 36 个显著差异表达 mRNAs(DEmRNAs)(图 4E)。通过关键驱动基因分析(Key Driver Analysis, KDA),从 36 个 DEmRNAs 中鉴定出 10 个关键调控基因,这些基因在 OP 和 OR 小鼠的基因调控网络中起核心作用(图 4F,表 S7)。关键驱动基因中表达最显著的基因:Fbln2、Col6a3、Col3a1、Col1a1 和 Bgn 在 OR 小鼠中的表达显著高于 OP 小鼠,p 值分别为 8e-8、4.17e-5、1.50e-4、2.18e-4 和 9.20e-4。
通过免疫组化(IHC)检测白细胞介素-6(IL-6)的组织表达来评估结肠炎症状态(图 5A)。采用 H-score 评分系统量化 IL-6 免疫反应性,结果显示 OP 组的 H-score 显著高于 OR 组(p < 0.05)(图 5B),表明 OP 小鼠的炎症反应更明显。定量实时 PCR(qRT-PCR) 验证 10 个关键驱动基因的表达水平,结果显示:其中 8 个基因在 OR 小鼠中显著上调(p < 0.05)(图 5C)。
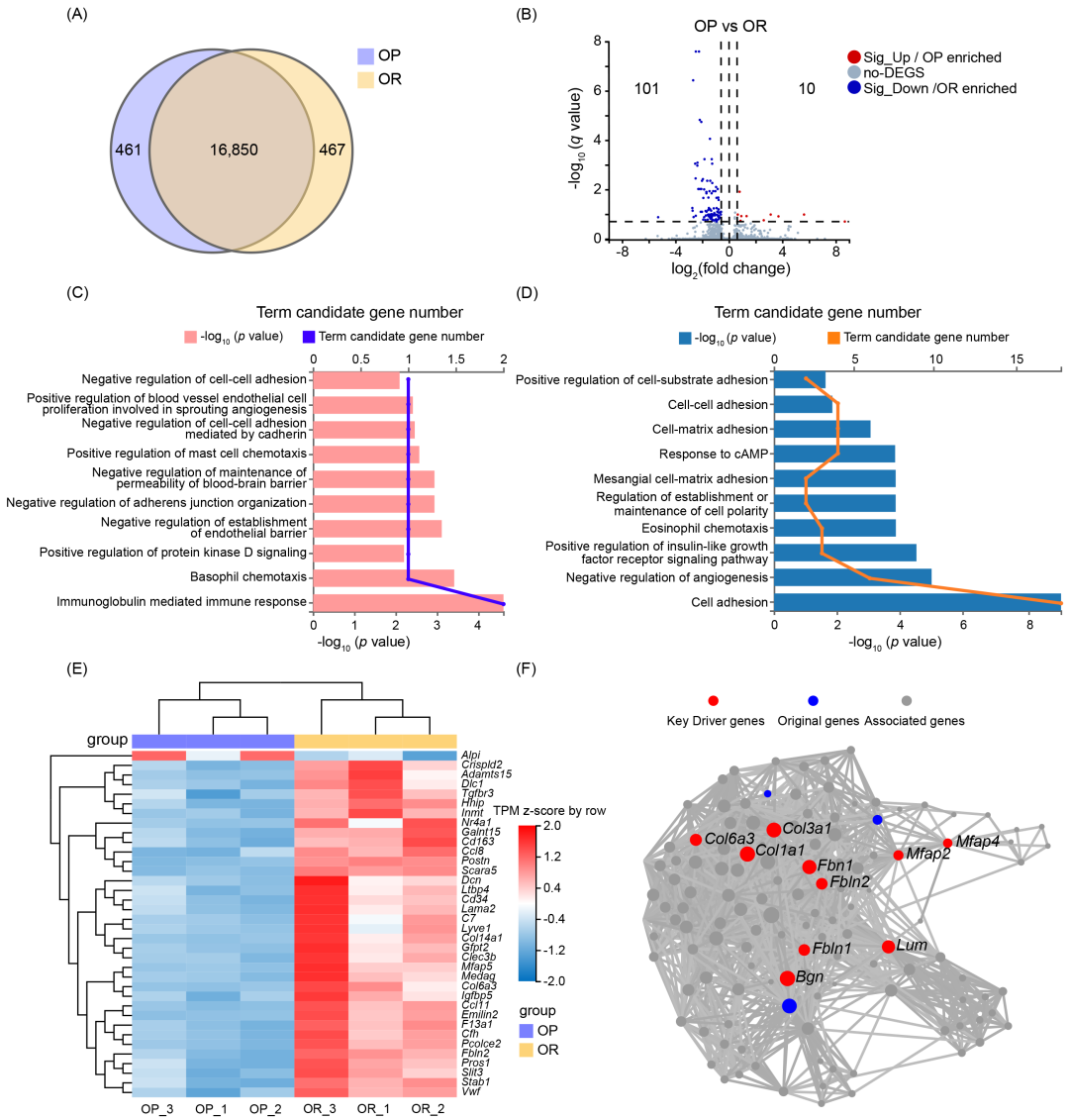
图4. OP 和 OR 小鼠结肠 RNA-seq 分析
(A) Venn 图 显示 OP 组和 OR 组基因数量的分布。两组小鼠共有 16,850 个共同基因。OP 组特有 461 个基因,OR 组特有 467 个基因。(B) 火山图(Volcano plot) 展示 OP 和 OR 小鼠差异表达基因(DEGs) 的分布。判定标准:fold change ≥ 1.2 或 ≤ 0.83,q-value < 0.2。(C, D) 基因本体论(GO)功能富集分析,展示差异表达基因的主要生物学功能。(E) 层次聚类分析 显示 36 个显著差异表达基因 在 OP 和 OR 小鼠中的表达模式。判定标准:fold change ≥ 1.2 或 ≤ 0.83,q-value < 0.05。(F) 关键驱动基因(Key Driver Genes)的可视化网络。节点的大小代表该基因在基因调控网络中与其他基因的连接数量(即其在网络中的关键性)。
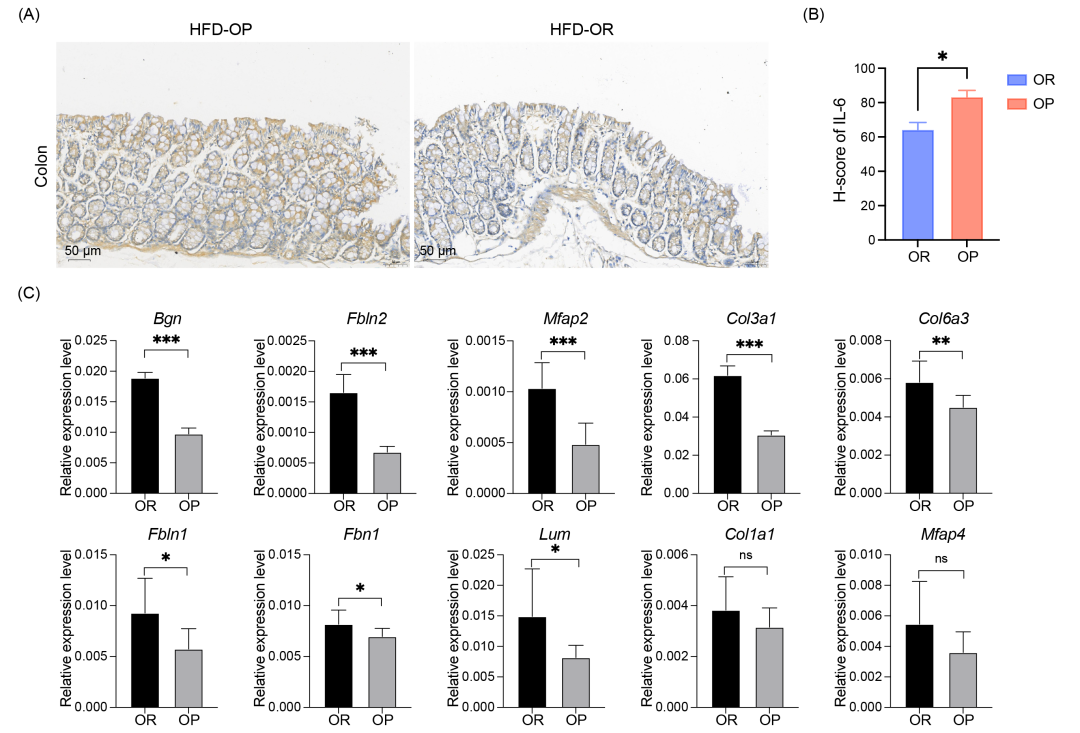
图5. 基于转录组数据验证肠道屏障反应
(A) 通过免疫组化分析检测结肠组织中白细胞介素-6(IL-6)的表达。(B) IL-6 免疫反应性 H-score 在 OP 组 和 OR 组中的比较。统计分析:非配对,双尾 t 检验,n = 3,*p < 0.05。(C) qRT-PCR 验证 OP 和 OR 小鼠的 10 个结肠屏障驱动基因。统计分析:非配对,双尾 t 检验,n = 3,*p < 0.05;**p < 0.01;***p < 0.001。
微生物组、代谢组和转录组之间的相关性
肠道微生物与代谢物的相关性通过肠道优势菌属与潜在代谢物的相关性分析,共鉴定出 20 种肠道细菌属与 32 种代谢物 之间的相关性。Kineothrix、Butyricicoccus、Intestinimonas、Rumiclostridium 和 Faecalicatena与氨基酸代谢物呈负相关,与胆汁酸代谢物呈正相关。Longibaculum、Butyricicoccus、Intestinimonas、Rumiclostridium 和 Faecalicatena与氨基酸代谢物呈正相关,与胆汁酸代谢物呈负相关(图 6A)。
粪便代谢物与关键驱动基因的相关性研究分析了与细胞粘附、迁移及细胞外基质结构相关的驱动基因(Driver Genes)与粪便代谢物的相关性(图 6B)。胆汁酸代谢物 与肠道关键驱动基因(Col6a3、Fbln1、Bgn 和 Mfap2)呈正相关。氨基酸代谢物与肠道黏膜屏障相关驱动基因(Col6a3、Fbln2、Col1a1、Col3a1、Fbln1、Bgn 和 Mfap2)呈负相关。
肠道微生物与关键驱动基因的相关性分析得到,不同属级肠道微生物与细胞粘附、迁移及细胞外基质结构相关基因之间的相关性如图 6C所示。Longibaculum 与 Col6a3 显著负相关。Traorella、Fournierella 和 Rumiclostridium 显著正相关 (Fbn1、Lum、Bgn、Mfap2、Col6a3、Fbln2、Col1a1 和 Col3a1)。
对血清和粪便代谢组进行了整合分析,发现:粪便代谢组中 42 条 KEGG 代谢通路发生改变。血清代谢组中 21 条 KEGG 代谢通路发生改变(图 6D)。6 条氨基酸代谢相关通路在 OP 小鼠中富集程度高于 OR 小鼠。2 条消化系统相关通路在 OR 小鼠中的富集程度低于 OP 小鼠。粪便和血清代谢组共有 16 条 KEGG 代谢通路(表 S9)。
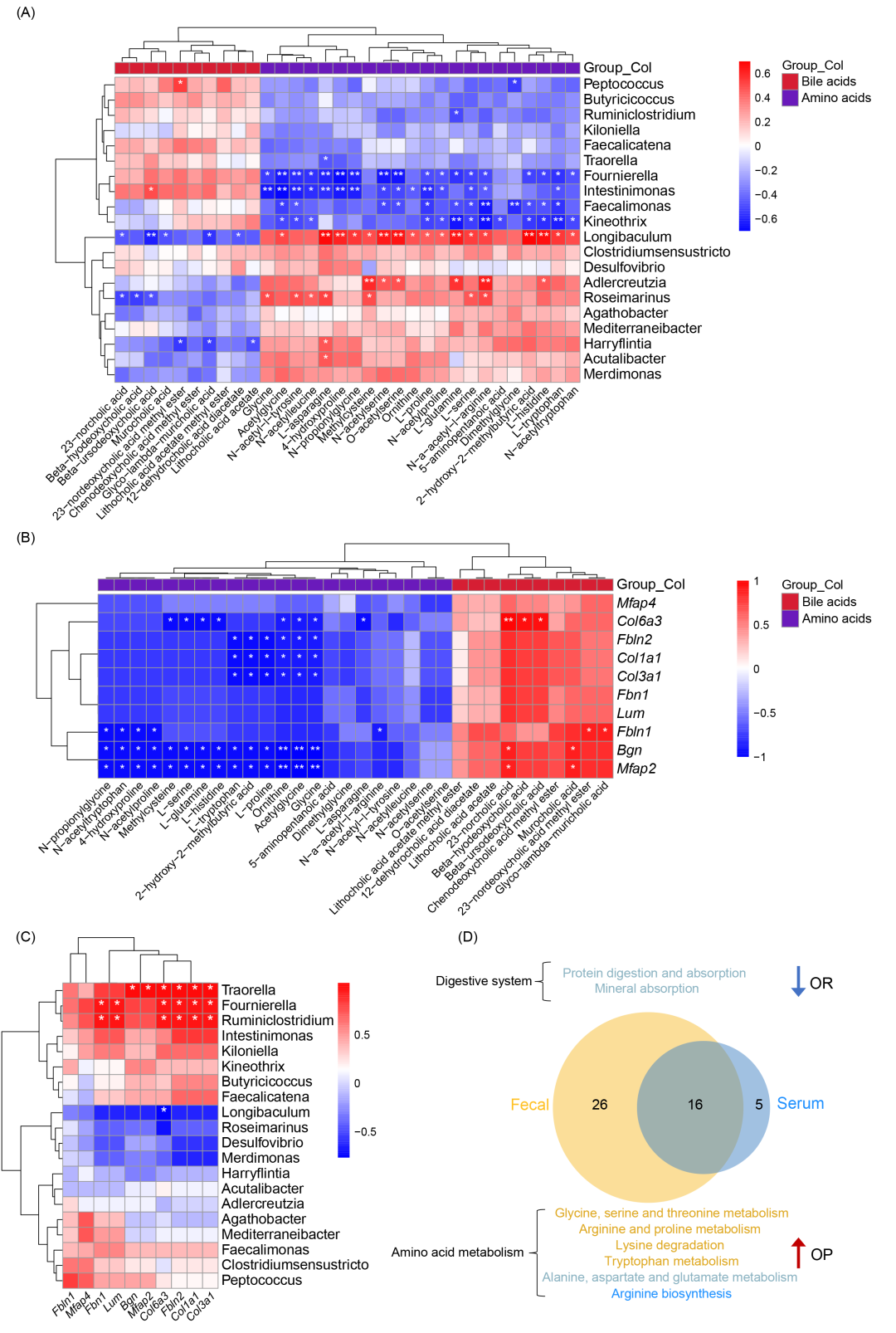
图 6. 微生物组、代谢组和转录组之间的相关性分析
(A) 肠道微生物与粪便中差异代谢物的Spearman相关性分析。X轴和Y轴分别为差异代谢物和菌属。p < 0.05 标记为 “*”,p < 0.01 标记为 “**”。(B) 代谢组与转录组的联合分析。X轴和Y轴分别为粪便中的差异代谢物和驱动基因。p < 0.05 标记为 “*”,p < 0.01 标记为 “**”。(C) 菌属水平肠道微生物与 10 个关键驱动基因的 Spearman 相关性分析。X轴和Y轴分别为驱动基因和差异菌属。p < 0.05 标记为 “*”,p < 0.01 标记为 “**”。(D) OP 和 OR 组 GO 术语数量的 Venn 图。通路名称的颜色与 Venn 图中所代表的组别颜色一致,即:黄色通路代表粪便组;蓝色通路代表血清组;绿色通路代表两组重叠的通路。
讨 论
我们通过微生物组测序、代谢组学和转录组学分析,研究了肥胖易感(OP)和肥胖抵抗(OR)表型差异与宿主-微生物组相互作用之间的关系,如图示摘要所总结。我们的研究表明,Longibaculum 和 Kineothrix 可能分别作为 OP 和 OR 小鼠肠道微生物群动态变化的关键菌属,可能引发两组微生物群体的社区效应。有益微生物群的增加、非 12-OH 胆汁酸代谢物的增加以及肠道屏障的强化与抗肥胖表型显著相关。Longibaculum-UDCA-Col6a3 通路可能是其潜在的作用机制。肠道黏膜屏障的损伤和细菌移位可能加剧体重增加,而氨基酸谱的差异或升高可能作为肥胖易感代谢表型的预测指标。
我们首先分析了 OP 和 OR 小鼠肠道微生物群的动态变化,并确定了核心优势菌属。值得注意的是,肥胖表型差异表现为多菌群的参与,各种肠道微生物因素和代谢失调共同导致宿主-微生物相互作用的异常。然而,目前尚无关于小鼠 OP 和 OR 形成过程中微生物组变化的报道。我们的研究揭示了 Longibaculum 和 Kineothrix 分别是 OP 和 OR 小鼠的关键菌属。特别地,Kineothrix 属在 OR 小鼠和 OR 人群中均显著升高。属于 Kineothrix 属的物种 Kineothrix alysoides在脂肪肝动物模型中治疗后显著增加57倍,并改善了肠道屏障功能。我们的转录组学结果显示,与 OP 小鼠相比,OR 小鼠的肠道细胞粘附功能通路显著富集,包括细胞粘附的正向调控和细胞-基质粘附。
此外,经过 14 和 16 周的高脂饮食喂养后,其他稳定存在于 OR 小鼠中的细菌属包括Butyricicoccus、Intestinimonas、Rumiclostridium和Faecalicatena,这些菌属与小鼠或人类体重减轻相关的微生物改变一致。这四种有益细菌(Butyricicoccus、Intestinimonas、Rumiclostridium和 Faecalicatena)的功能与 Kineothrix 相似,对短链脂肪酸(SCFAs)的生产具有重要贡献。Butyricicoccus 作为一种产丁酸的细菌,与其他有益细菌协同作用,降低了 Firmicutes 和 Bacteroidetes(F/B)比率。值得注意的是,我们的研究发现,OR 小鼠的 F/B 比率显著低于 OP 小鼠,这表明 OR 小鼠的肠道稳态优于 OP 小鼠(图 S6)。OP 小鼠 F/B 比率的显著增加被认为是肠道微生物群失,促进脂质生产、异常体重增加和慢性代谢疾病的发生。Butyricicoccus 作为一种保护肠道屏障的细菌,在维持肠道稳态中起着至关重要的作用,通过确保粘液的产生和维持紧密连接的完整性。口服 Butyricicoccus 可以减少结肠炎大鼠的肠道炎症。在肥胖患者中,Butyricicoccus 的高丰度有助于体重减轻期间体质指数(BMI)的降低,这表明其在促进抗肥胖代谢表型中的潜在作用。Intestinimonas,我们在 OR 代谢表型中也发现其高表达,在肠道内将氨基酸转化为丁酸。我们发现,OR 小鼠中 22 种氨基酸显著减少,并且多种氨基酸与 Intestinimonas 显示显著的负相关,表明其在调节蛋白质平衡和维持肠道稳态中的关键作用。补充剂缓解了 OP 小鼠中 Intestinimonas 丰度减少和肠道微生物群失调的状态,从而改善了 OP 小鼠的代谢状态。Rumiclostridium 与大多数代谢参数(如肠道通透性、血脂、葡萄糖和胰岛素)呈强负相关。为了维持肠道免疫和机械屏障的关键驱动基因与 Rumiclostridium 在 OR 小鼠中的上调表现出显著正相关。此外,Rumiclostridium 参与调节脂质代谢和促进白色脂肪组织中的褐色脂肪细胞发育,暗示其通过影响能量代谢代谢物减少体重。所有这些稳定存在的群落都与 OR 表型相关,基本上都生产 SCFAs,这对维持肠道上皮细胞功能和结构有益,同时调节肠道微生物群的平衡。这些结果表明,OR 小鼠的抗肥胖表型可能是由肠道微生物群的社区效应塑造的。因此,OR 小鼠的表型与有益的肠道共生菌密切相关,即Butyricicoccus、Intestinimonas、Rumiclostridiu和 Faecalicatena,以 Kineothrix为主。
相比之下,OP 小鼠中稳定存在的关键菌属 Longibaculum 与维持肠道机械屏障的驱动基因 Col6a3 以及肠道黏膜完整性指数表现出显著的负相关。属于该属的 Longibaculum muris 在高蛋白饮食小鼠中的丰度增加,其粪便水平与大鼠口服葡萄糖耐受不良呈正相关。然而,Longibaculum 影响其功能的潜在通路尚不清楚。我们观察到,其他与 Longibaculum 稳定共存的细菌属(如 Harryflintia、Adlercreutzia 、Agathobacter和 Roseimarinus)也参与了 OP 表型的形成,这些细菌变化与之前关于超重小鼠和人类的微生物改变一致。这些有害细菌(Harryflintia、Adlercreutzia 和 Agathobacter)与代谢标志物(如体重、炎症、脂肪量和葡萄糖水平)呈正相关,这些标志物与肥胖及相关代谢紊乱密切相关。此外,Harryflintia 与某些与肠道屏障完整性和粘液功能相关的标志物表现出负相关。Adlercreutzia 与体重的正相关性是前瞻性的, 表明其与肥胖之间存在双向因果关系。Agathobacter 的比例与 1‒2.5 岁儿童超重和肥胖的快速发展呈正相关。尽管关于 Roseimarinus 与肥胖的关系研究较少,但它可能作为肥胖的新标志物。这些细菌属的丰度增加是肥胖相关失调的共同特征,表明它们可能作为有害微生物群,促进 OP 表型的形成。我们推测,OP 表型与有害肠道共生菌密切相关,即Agathobacter、Roseimarinus、Harryflintia 和Adlercreutzia,以 Longibaculum为主。因此,提示肠道有益细菌的增加可能抵消高脂饮食后的肥胖,而肠道微生物群的失调、代谢异常和肠道黏膜屏障的削弱则加剧体重增加。
肠道微生物群通过厌氧发酵未消化的饮食成分以及来自微生物和宿主的内源性物质产生各种代谢物。这些微生物代谢物可以穿透宿主细胞,直接与上皮细胞和黏膜屏障相互作用,或者进入门静脉血液,影响全身的代谢健康。我们的研究通过将代谢组学与微生物组测序分析结合,揭示了肠道微生物群与代谢物之间的强相关性,表明胆汁酸、氨基酸、短链脂肪酸、吲哚衍生物和其他代谢物水平的变化可能是 OP 和 OR 表型差异的潜在基础。
我们的研究发现,与 OP 小鼠相比,OR 小鼠中非 12-OH 胆汁酸(Muricholic acid [MCA]、UDCA、HDCA、Lithocholic acid [LCA]、Chenodeoxycholic acid [CDCA] 和其他替代途径衍生物)显著升高。此外,通过 PICRUSt 分析的 16S rDNA 基因测序结果显示,OR 小鼠中胆汁酸代谢通路显著富集(图 S7)。CYP7B1,作为胆汁酸替代途径中的关键酶,在 OR 小鼠中显著上调,表明胆汁酸替代途径的激活,同时与 胆汁盐水解酶(BSH)基因的高丰度相关。在本研究中,OR 小鼠中的 BSH 丰富细菌(如 Bacteroides、Lactobacillus、Bifidobacterium 和 Clostridium)被抑制,导致远端结肠中结合胆汁酸的积累,这些胆汁酸主要是非 12-OH 胆汁酸。Longibaculum 也属于具有 BSH 基因的属(https://www.ncbi.nlm.nih.gov/nuccore/2428088975)。这些发现表明,肠道微生物群失调通过调节替代胆汁酸途径对抗体重增加起到了重要作用。此外,我们的干预研究表明,含有 UDCA 或 HDCA(非 12-OH)补充剂的高脂饮食抑制了小鼠的体重增加,进一步确认了替代胆汁酸合成途径的作用。已有研究报告表明,UDCA 或 HDCA 补充剂可通过恢复肠道屏障完整性减少代谢功能障碍。此外,我们的分析揭示了 UDCA 和 Col6a3(肠道屏障完整性的关键驱动基因) 显著正相关,而 UDCA 和 Longibaculum 显著负相关,表明通过抑制含有 BSH 基因的细菌(如 Longibaculum),激活次级胆汁酸(如 UDCA),通过上调涉及细胞粘附和迁移的基因(如 Col6a3)来维持肠道屏障稳定性,并有助于减缓体重增加。
氨基酸谱是肥胖易感性的潜在生物标志物,并被认为是儿童肥胖早期干预的有用标志物。然而,尚无进一步的动物研究。当前的研究确定了 22 种肠道氨基酸代谢物和 6 条氨基酸代谢通路在 OP 小鼠中的显著过度表达;氨基酸水平的升高与肠道微生物群失调相关。Bacteroides vulgaris 被确定为介导特定氨基酸生物合成与胰岛素抵抗之间联系的主要物种。胰岛素抵抗个体的血清代谢组表现出特定氨基酸的浓度升高。高纤维饮食后胰岛素抵抗和葡萄糖代谢的改善也与Prevotella-to-Bacteroides 比率的增加相关。富含 Prevotella 的肠道微生物群有助于减重和减少胆固醇。我们的研究支持这一假设。Prevotellaceae、Prevotellamassilia、Prevotella/Bacteroides 比率、肠道氨基酸谱的过度表达和6 条氨基酸通路的表达抑制反映了 OR 小鼠的代谢表型(图 S6)。肠道中过多的氨基酸也会影响肠道微生物群,促进 Bacteroides、Clostridium 和Helicobacter pylori 等有害细菌的生长。这表明,氨基酸失衡与肠道细菌的相互作用显著促进了肥胖易感代谢状态的形成。此外,在一项为期 12 年的随访研究中,Wang 等人强调了氨基酸代谢在糖尿病早期发病中的潜在重要性,并建议氨基酸谱有助于糖尿病风险评估,并可能作为未来无糖尿病个体糖尿病的预测因子。其他研究表明,特定氨基酸的血浆水平与 BMI和葡萄糖抵抗之间存在显著正相关。我们的研究强调了氨基酸谱作为肥胖易感性标志物的潜力,体现在 OP 小鼠血液和粪便中氨基酸代谢通路的过度表达。因此,我们的研究表明,肥胖相关氨基酸的失衡或增加可能加剧 OP 表型,而肥胖相关胆汁酸代谢的维持则支持 OR 表型,肠道微生物群在这一调节过程中起着关键作用。
肠道屏障功能包括机械、生态和免疫三大屏障。生态屏障的失调,以微生物群失调为特征,通常会导致机械和免疫屏障的功能障碍,并引发炎症。为研究肠道微生物群与宿主免疫和肠道屏障反应之间的潜在关联,我们对 OP 和 OR 小鼠的结肠样本进行了 RNA-seq 分析。结肠是肠道中微生物群最密集、代谢活性最强的区域,包含超过 1013 个微生物细胞。我们研究中的 GO 分析表明,OP 小鼠肠道细胞粘附功能受到破坏,肠道屏障的完整性受损使得共生微生物能够接触肠道上皮细胞。在肠道屏障受损的情况下,OP 表型可能与潜在病原微生物和/或细菌群体组分的增加相关,如 Bacteroidetes、Clostridium 和 Helicobacter pylori,并触发慢性炎症反应。通过免疫组化分析,我们确认了 OP 小鼠中炎性细胞的增加。根据 GO 富集结果,与细胞粘附、迁移及细胞外基质结构相关的关键驱动基因(Col6a3、Bgn、Fbln2、Mfap2、Col3a1、Fbln1、Fbn1 和 Lum)在 OR 小鼠中显著上调,且通过实时 PCR 验证了这一结果。大多数先前研究报告了这些基因在肥胖小鼠或人类脂肪组织中的表达下降,我们的研究确认了 OP 小鼠结肠中这些基因的表达下降。总结来说,肠道黏膜屏障受损和细菌移位可能加剧体重增加,而 OR 小鼠的肠道稳态和屏障完整性得到了维持。
我们的研究聚焦于宿主基因表达在结肠中的调节作用,结肠是肠道中微生物群最丰富、最多样化的地方。结肠是复杂发酵过程的核心,生产如胆汁酸衍生物、氨基酸代谢物和短链脂肪酸(SCFAs)等代谢物。这些方面是我们代谢组学和微生物组分析的核心,为进一步的研究提供了宝贵的资源,例如粪便微生物群移植实验和基于细胞的研究。然而,不同肠道区域在微生物组成和代谢物谱方面存在显著差异,强调了未来需要探索整个胃肠道的代谢组学和微生物组学。
结 论
我们的结果表明,关键菌属可能通过菌群效应影响微生物群动态变化的改变。肠道黏膜屏障的损伤、有害菌富集以及肥胖相关氨基酸水平的升高可能加剧体重增加。相反,OR 小鼠的肠道微生物群能够维持稳态,升高非 12-OH 胆汁酸并维持肠道屏障的完整性。Longibaculum-UDCA-Col6a3 通路的重要性也得到了强调。这些发现有助于识别肥胖相关的关键肠道微生物标志物和潜在的抗肥胖靶点,为推动对肥胖易感和抵抗表型的理解提供了宝贵的资源。
方 法
动物实验设计
总共购买了 87 只雄性 C57BL/6J 小鼠(4 周龄,17‒19 克),由上海斯莱克实验动物有限公司提供,并在浙江大学动物实验中心适应 1 周。其中,47 只小鼠(n = 47)被随机分配到两组饮食:正常饮食组(13% 脂肪,n = 5,江苏协同药业生物工程有限公司提供)。高脂饮食(HFD)组(60% 脂肪,D12492,n = 42,Trophic Animal Feed 高科技有限公司提供)。并喂养 16 周。小鼠在 0、2、5、14 和 16 周(分别对应 5、7、10、19 和 21 周龄)采集粪便样本,以进行肠道微生物组基因组分析。代谢组学分析基于 16 周喂养期的小鼠粪便样本。在 16 周的喂养期或 21 周龄时,将小鼠麻醉(腹腔注射 1% 戊巴比妥钠),并采集约 0.5‒1 毫升血液。通过离心(3000 rpm,10 分钟)分离血清。处死后采集肝脏、结肠和白色脂肪组织样本。组织样本保存在 -80°C 直至分析,另一部分组织样本则用组织固定液固定并石蜡包埋保存。
为了检测 HDCA 和 UDCA 补充对体重的影响,另外 40 只小鼠 随机分配到四组饮食:正常饮食组(n = 10)(13% 脂肪),高脂饮食组(n = 10)(60% 脂肪),高脂饮食 + 0.5% UDCA 组(n = 10)(60% 脂肪),高脂饮食 + HDCA 组(n = 10)(60% 脂肪)。实验期间,每周监测小鼠体重。小鼠在 8 周喂养期或 12 周龄时麻醉,并收集结肠组织。所有小鼠均在特定病原体自由条件下饲养,提供自由取食和饮水,环境条件控制在 温度 20‒22°C,湿度 45 ± 5%,12 小时光照/暗周期。本研究获得了浙江大学伦理委员会的批准,并遵循浙江大学实验动物中心的相关指南(批准号:ZJU20220424)。
OP和OR小鼠的定义
在 16 周喂养期或 21 周龄时,HFD 喂养组中的 42 只小鼠根据高脂饮食喂养期间的体重增长分为两组:体重增长排名前 10 的小鼠被定义为 OP(上四分位),体重增长排名后 10 的小鼠被定义为 OR(下四分位)。其余的 22 只小鼠被排除在分析之外。
人群研究设计
研究参与者从 2019 年 6 月至 8 月期间在浙江省兰溪市的城市地区开展的兰溪队列社区研究中招募。参与者的纳入和排除标准的详细信息见补充信息方法 S1。总共有 586 名高能量摄入的参与者被纳入。BMI ≥ 24 kg/m² 的参与者被定义为 OP 组(n = 248),BMI < 24 kg/m² 的参与者被定义为 OR 组(n = 338)。通过面对面的问卷调查和体格检查获取基线信息,包括性别、年龄、吸烟和饮酒状况、教育水平、体脂百分比、身体活动水平和生化数据。每位参与者的粪便样本在队列调查当天也被采集并保存在 -80°C 直至分析。连续变量以均值和标准差(SD)或标准误差(SEM)表示。分类变量以百分比(%)表示。本研究已获得浙江大学公共卫生学院伦理委员会的批准(ZGL201905-1)。
粪便样本中的微生物组测序和数据分析
DNA 提取
从粪便样本中使用DNeasy® PowerSoil® Kit(Qiagen,德国希尔登)按照制造商的说明提取总基因组 DNA。随后,使用Qubit和 1% 琼脂糖凝胶电泳分别检测提取的基因组 DNA 的浓度和完整性。
PCR 扩增和微生物组测序
使用引物341F (5ʹ-CCTACGGGNGGCWGCAG-3ʹ)和805R (5ʹ-GACTACHVGGGTATCTAATCC-3ʹ)(结合条形码序列)扩增细菌 16S rDNA 基因的 V3‒V4 区域。制备扩增子池进行测序,并使用 Agilent 2100 Bioanalyzer 和 Illumina 文库定量试剂盒 评估扩增子库的大小和数量。文库使用 NovaSeq PE250 平台 进行测序。
数据分析
我们使用 R 语言分析管道(由 Liu 等提出)将原始读取数据(fastq 格式)转化为 扩增子序列变异(ASVs)表。ASVs 是具有 100% 相似性的序列。ASV 丰度矩阵和注释结果为随后的生物信息分析提供基础,包括多样性分析(α、β)、物种组成和 LEfSe 分析,分析对象为 OP 和 OR 小鼠以及 OP 和 OR 人群。采用 Mann-Whitney U 检验,对组间的 α 多样性指数(Shannon 和 Simpson 指数)进行比较。为分析不同组之间的 β 多样性,使用 Bray-Curtis 距离作为度量标准,执行主坐标分析。为分析两组之间的差异,使用 LEfSe 分析,设定 p < 0.05 和 LDA 评分 > 2.0。使用 Spearman 等级相关系数计算细菌、代谢物和基因之间的相关性。此外,为 OP 和 OR 人群组进行 多变量线性回归 和 Permanova 分析,以调整性别、年龄、吸烟状况、饮酒、教育水平和体脂百分比等因素。
血清和粪便样本中的代谢组测量和数据分析
血清样本准备
血清样本在冰上解冻,使用含有内标 1 的提取剂(甲醇:乙腈:水 = 4:2:1,v/v)和 50% 甲醇缓冲液提取血清代谢物。所有样本通过液相色谱-质谱联用(LC-MS)进行分析,使用 Waters 2777C UPLC 系统(Waters,MA)和 Q Exactive HF 高分辨率质谱仪(Thermo Fisher Scientific,MA),根据制造商的说明进行操作。
粪便样本准备
从冰上解冻的粪便样本中提取肠道代谢物,使用 50% 甲醇缓冲液提取。样本、质量控制(QC)和标准品通过 LC-MS 及样本稀释液分析。使用 Waters ACQUITY UPLC I-Class Plus(Waters,MA)与 QTRAP6500 Plus 高灵敏度质谱仪(SCIEX,MA)进行代谢物分离和定量。每个多反应监测(MRM)转化(离子转化)通过 HMQuant 软件 识别和整合。
数据分析
使用 MetaX 软件 对仪器采集的数据进行预处理,然后使用 人类代谢组数据库 和 KEGG 进行代谢物的分类特征和功能属性分析。首先对数据进行 log2 转换,建立实验组之间的 偏最小二乘-判别分析(PLS-DA) 模型,并采用 Pareto 方法 进行标准化。应用 正交信号校正(OPLS-DA) 进行实验组之间的分析,减少模型复杂性并增强其解释能力,同时确保预测能力。计算 VIP 值,评估每个代谢物表达模式对样本分类判别的影响和解释能力,VIP > 1 的代谢物被认为对样本分类有帮助。差异分析还包括常规的单变量分析方法,如 fold-change 分析 和 t 检验。使用 Welch’s t 检验 测试各组之间每个代谢物表达水平的显著性,获取 p 值,并使用 Benjamini‒Hochberg 算法 对 p 值进行校正,得到 q 值。q 值用于评估两组样本之间的显著性差异。 fold change 反映了两组样本中代谢物的平均值变化,q 值则反映该变化是否具有统计学意义。对于 fold change ≥ 1.2 或 ≤ 0.83 且 p 值 < 0.05 的代谢物,认为它们显著不同。对差异代谢物进行 受试者工作特征(ROC)曲线分析,在 ROC 分析中,AUC > 0.75 表示该代谢物作为生物标志物的能力。
结肠样本的转录组 RNA 测序和数据分析
使用 TRIzol 试剂(Invitrogen,CA)提取和纯化总 RNA,按照制造商的说明操作。mRNA 从总 RNA 中富集,并使用 DNBSEQ 高通量平台构建并测序特异性转录组文库。原始测序数据使用 SOAPnuke(v1.5.6)过滤,以获得清洁数据。数据挖掘使用 Dr Tom 的多组学数据挖掘系统(https://biosys.bgi.com)。使用 Bowtie2(v2.3.4.3)将清洁数据与参考基因集(GCF_000001635.27_GRCm39)比对进行差异基因分析。使用 RSEM(v1.3.1)软件量化基因表达,使用 pheatmap(v1.0.8)生成不同样本中的基因表达热图。使用 DESeq2(v1.4.5)检测 OP 和 OR 小鼠之间的差异表达基因,判定标准为 fold-change ≥ 1.2 或 ≤ 0.83 且 q 值 < 0.2。为进一步探索与表型变化相关的基因功能,使用 GO(http://www.geneontology.org)和 KEGG(https://www.kegg.jp/)对差异表达基因进行富集分析,采用 Phyper(https://en.wikipedia.org/wiki/Hypergeometric_distribution)进行超几何测试,q 值 ≤ 0.05 作为候选基因显著富集的阈值。为识别选定基因中的关键驱动基因,基于 STRING11 数据库 的分数进行 关键驱动基因分析(KDA),截止点为 > 500(BGI Dr.Tom 系统)。
H&E 染色
用于组织学分析时,白色脂肪组织和肝组织被固定在 4% 甲醛中,石蜡包埋,切片并用 H&E 染色。使用显微镜(Olympus,东京,日本,100 × 放大)评估脂肪小泡的大小、炎症程度以及肠道和隐窝损伤的严重性和范围。每个切片的图像被拍摄并使用 ImageJ 1.54g 软件(美国国立卫生研究院)定量测量 eWAT 细胞和肝细胞的空泡区域。
免疫组化(IHC)染色和评分
结肠组织切片首先在 二甲苯去蜡,然后使用逐级乙醇系列重新水化。在消耗内源性过氧化物酶并阻断抗原后,切片与IL-6 的一抗(1:200,Servicebio,中国)孵育过夜,温度 4°C。随后,切片与二抗(AiFang Biological,中国)室温孵育 30 分钟。染色使用 二氨基联苯胺显色,并用 苏木精 进行对比染色。染色后的切片使用 光学显微镜(Nikon E100 DS-U3 相机系统,东京,日本) 进行成像。微观图像使用 Image-Pro Plus 6.0 软件进行分析。IL-6 免疫反应性通过 H-score 量化,H-score 计算公式为:H-score = ∑(pi × i),其中 pi 为阳性细胞的百分比,i 为特定染色的强度。特定染色的相对强度定义为:负(值 = 0),低阳性(值 = 1),阳性(值 = 2),高阳性(值 = 3)。
qRT-PCR 分析
使用 LightCycler 480 仪器(罗氏,巴塞尔,瑞士)进行 16S rRNA 定量 PCR。所有反应在一个运行中重复进行,并且每个 PCR 运行中重复两次。样本在 10 μL 反应混合液中分析,包含 6.5 μL 1× SYBR green master mix 缓冲液(q311-02,Vazyme,中国),每对引物浓度为 0.2 μM,1 μL 来自粪便提取的基因组 DNA。标准曲线使用 Kineothrix_alysoides 和 Longibaculum_muris 的 16S rRNA PCR 产品。
代码和数据可用性
高脂饮食(HFD)诱导的 C57BL/6J 小鼠 的 16S rRNA 扩增子数据 已上传至 NCBI SRA 数据库,Bioproject 编号为 PRJNA1048179(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1048179)。高脂饮食诱导的 C57BL/6J 小鼠结肠中的 mRNA 表达谱 已上传至 Gene Expression Omnibus (GEO) 数据库,获取编号为 GSE249390(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE249390)。本研究报告的人类原始序列数据已上传至 Genome Sequence Archive (GSA),获取编号为 CRA017756(https://ngdc.cncb.ac.cn/gsa/s/LgGtbDZv),由 中国国家基因组数据中心(中国生物信息学中心/北京基因组研究所) 托管。使用的数据和脚本保存在 GitHub(https://github.com/candice1029/OPOR-project/tree/main/OPOR)。附加材料(方法、图表、图形摘要、幻灯片、视频、中文翻译版本及更新材料)可通过在线 DOI 或 iMeta Science 查阅(http://www.imeta.science/imetaomics/)。
引文格式:
Congcong Wang, Jinhua Lin, Meng Duan, Jialing He, Simayi Halizere, Ningxin Chen, Xinyu Chen, et al. 2025. “Multi-omics reveals different signatures of obesity-prone and obesity-resistant mice.” iMetaOmics 1: e59. https://doi.org/10.1002/imo2.59.
作者简介
王聪聪(第一作者)
● 硕士毕业于浙江大学公共卫生学院,德国亥姆霍兹慕尼黑研究中心在读博士生。
● 研究方向为代谢相关疾病、代谢生理机制及多组学研究,以第一作者在iMetaOmics、iScience、Metabolites等期刊上发表多篇研究论文。

林锦华(第一作者)
● 硕士研究生。2024年6月毕业于浙江大学公共卫生学院公共卫生专业。
● 研究方向为肥胖及肥胖相关疾病研究,在iScience、The journal of nutrition, health & aging等期刊发表SCI论文2篇。

段萌(第一作者)
● 博士研究生。2024年6月毕业于浙江大学公共卫生学院营养与食品卫生学专业。
● 研究方向为肥胖及肥胖相关疾病研究,以共同第一作者或参与者在iScience、Cancer letters、Clinical Nutrition、Nature Communications等期刊发表多篇研究论文。

杨菲(通讯作者)
● 浙江大学医学院公共卫生学院副教授,博士生导师。
● 研究方向为肥胖及肥胖相关疾病影响因素及机制研究。主持国家自然科学基金2项,作为骨干参加国家重点研发计划1项。以第一/通讯作者(含共同)在Cell Metabolism、Cancer Letters、Cellular & Molecular Immunology、Cell Research、The Journal of Immunol、Cell Death & Disease、iScience、BMJ Open、Metabolites、European Journal of Immunology等杂志发表多篇研究论文。

朱善宽(通讯作者)
● 浙江大学求是特聘教授,二级教授,博士生导师,中华医学基金会(CMB)杰出教授,多伦多大学教授(Status Only),国务院政府特殊津贴学者,国家重点研发计划首席科学家,2014-2023连续十年入选爱思唯尔(Elsevier)中国高被引学者。
● 研究方向为慢性病流行病学,营养流行病学,人群-基础(P to B)转化,多组学研究等。已发表学术论文130余篇,SCI总被引超过2万次,Google Scholar总被引超过3万5千次。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics被DOAJ收录! (2025/1/27)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
● iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
● iMetaOmics | 上科大刘雪松组开发基于通路的肿瘤细胞鉴别工具TCfinder
● iMetaOmics | 中山大学刘鹏/邹宇田-整合人工智能实现HER2阳性乳腺癌精准管理
● iMetaOmics | 安徽农大李晓玉组-丛枝菌根真菌对玉米内生菌群的影响
● iMetaOmics | 徐涛/黄蓉/苏国海-急性冠脉综合征纵向多组学队列建设
● iMetaOmics | 通过整合宏组学促进人类与环境健康发展
● iMetaOmics | 苏州大学林俊组-揭示活性微生物及益生元/益生菌与关节炎联系
● iMetaOmics | 中国药科大学徐文波开发叶绿体基因组数据分析软件
● iMetaOmics | 清华刘晓组和复旦王久存组揭示特定细菌在皮肤老化中的作用
●iMetaOmics | 中南大学夏晓波团队揭示青光眼和SLE发病机制新关联
●iMetaOmics | 庐山植物园刘芬组揭示了自噬在植物-根微生物互作机制中的调控作用
●iMetaOmics | 杨瑞馥/袁静综述微生物组与“同一健康”的联系
●iMetaOmics | 同济/上海交大-开发支持群体分组分析的宏基因组测序综合分析软件
●iMetaOmics | 陈绍鸣-关于靶向NF-κB的潜伏逆转剂及其在HIV潜伏期的表观遗传和突变影响的评论
● iMetaOmics | 甘肃农大刘自刚组-强抗寒甘蓝型冬油菜的基因组组装和基因组特征解析
● iMetaOmics | 南京农大朱伟云组-外周血清素在结肠稳态中的作用
●iMetaOmics | 魏来/贾慧珏/何明光-多组学助力揭示塑造转录组的基因型-微生物组相互作用
● iMetaOmics | 徐州医科大学朱作斌组-微生物对寿命的调节:机制和治疗策略
● iMetaOmics | 白立景/邢凯组-解析脊椎动物肠道微生物多样性的影响因素
● iMetaOmics | 刘永鑫/陈同-用于食物微生物组成和时间序列研究的微生物组数据库FoodMicroDB
● iMetaOmics | 重庆大学王贵学组-肠道微生物细胞外囊泡在神经退行性疾病中的新作用及其治疗策略
● iMetaOmics | 四川大学王红宁组-解析产气荚膜梭菌的基因组宿主适应性
● iMetaOmics | 北京协和医院杨启文组-ramR基因突变增强免疫激活和依拉环素耐药性
● iMetaOmics | 香港中文苏奇组-抗菌多肽开发中的见解: 一个多学科视角的观察
● iMetaOmics | 上科大刘雪松组开发CD4 TCR特异性预测工具Pep2TCR
● iMetaOmics | 江苏省农科院植物细菌团队-解析中国梨火疫菌特征及溯源分析
● iMetaOmics | 基因组所刘永鑫组-易扩增子(EasyAmplicon):用户友好的扩增子测序数据分析指南
● iMetaOmics | 东京科学大学奥村学组-Hyena架构蛋白质语言建模
● iMetaOmics | 兰大南志标/段廷玉组-丛枝菌根网络影响邻近植物对病原菌的响应
● iMetaOmics | 汝之蜜糖彼之砒霜:源自益生菌LGG的研究证据
● iMetaOmics | 中国农大汪杰组-解析微塑料胁迫下玉米的分子响应
● iMetaOmics | 陈嘉莉/唐少军-奶源动物双歧杆菌乳亚种的膳食健康
● iMetaOmics | 西北农林曹阳春组综述艰难梭菌感染病理机制及饮食模式对其影响策略
● iMetaOmics | 宁波大学叶央芳组-解析驱动蟹肠道菌群稳定性的关键物种
● iMetaOmics | 中国农科院毕研亮组-精确消化道微生物群调节策略促进宿主健康
● iMetaOmics | 深圳大学李猛组-沉积物中古菌的多样性和代谢潜能
● iMetaOmics | 华东理工叶邦策组-多组学分析分枝杆菌侵染宿主过程
● iMetaOmics | 中国农业大学曹志军组-MicroRNA-微生物群互作调控宿主健康
● iMetaOmics | 中国农科院王秋霞组-微生物在土壤养分转换和作物营养代谢中的作用
● iMetaOmics | 军事医学研究院高月团队-提供高原心肌损伤新的治疗靶点
● iMetaOmics | 解放军总医院第一医学中心卫勃组-微生物与疼痛之缘
● iMetaOmics | 兰州大学刘鹏飞组-全球RNA病毒AMGs功能多样性
● iMetaOmics | 浙大袁长征组-解析MIND膳食模式的代谢组学特征及其与认知健康的关联
● iMetaOmics | 温州医科大李校堃院士团队-解析百合地黄汤重塑睡眠剥夺所致的肠-脑轴失衡
● iMetaOmics | 张磊/赵国屏院士团队-利用HTBGC-Finder识别人类肠道中水平转移的生物合成基因簇
● iMetaOmics | 海军军医大学潘炜华组-解析高原人类皮肤微生物组特征
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 1559
1559

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









