






点击蓝字 关注我们
微生物学研究者的宏蛋白质组学指南

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imt2.70031
● DOI: https://doi.org/10.1002/imt2.70031
● 2025年5月6日,国家蛋白质科学中心(北京)李乐园等在iMeta在线发表了题为“The Microbiologist's Guide to Metaproteomics”的综述文章。
● 本综述由宏蛋白质组学倡议(Metaproteomics Initiative, www.metaproteomics.org)撰写,介绍了宏蛋白质组学所必需的关键原理、系统阐述宏蛋白质组学的核心原理、前沿技术及关键分析流程,涵盖实验设计、样本制备、质谱技术、数据分析策略与统计方法等全链条研究要素,旨在为微生物组学和蛋白质组学研究人员提供实践指南。
● 第一作者:Tim Van Den Bossche● 通讯作者:李乐园(lileyuan@ncpsb.org.cn)
● 合作作者:Tim Van Den Bossche、Jean Armengaud、Dirk Benndorf、Jose Alfredo Blakeley-Ruiz、Madita Brauer、Kai Cheng(程凯)、Marybeth Creskey、Daniel Figeys、Lucia Grenga、Timothy JGriffin、Céline Henry、Robert LHettich、Tanja Holstein、Pratik DJagtap、Nico Jehmlich、Jonghyun Kim、Manuel Kleiner、Benoit JKunath、Xuxa Malliet、Lennart Martens、Subina Mehta、Bart Mesuere、Zhibin Ning、Alessandro Tanca、Sergio Uzzau、Pieter Verschaffelt、王靖、Paul Wilmes、Xu Zhang(张旭)、张新、李乐园,宏蛋白质组学倡议(The Metaproteomics Initiative)
● 主要单位:Department of Biomolecular Medicine, Faculty of Medicine and Health Sciences, Ghent University、医学蛋白质组全国重点实验室,北京蛋白质组研究中心,国家蛋白质科学中心(北京),北京生命组学研究所
亮 点
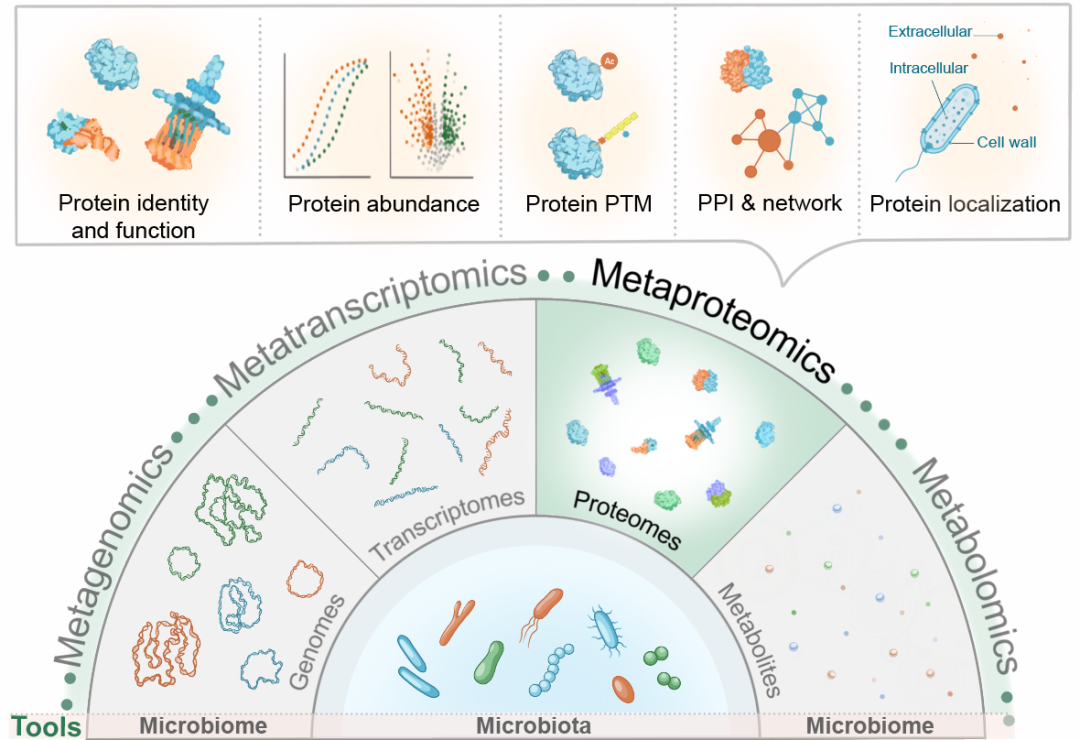
● 宏蛋白质组学可直接表征微生物群落的动态功能,其提供的蛋白质组层面解析为多组学研究提供了不可替代的功能维度补充;
● 本指南系统介绍了宏蛋白质组学研究中的实验设计、样本制备、质谱技术及数据分析等前沿方法;
● 本指南由宏蛋白质组学倡议(www.metaproteomics.org)组织编撰,旨在推动宏蛋白质组学技术普及,攻克领域内关键技术挑战,并推动跨领域合作以促进微生物组研究的创新发展。
摘 要
宏蛋白质组学是研究微生物组的新兴方法,能够在多样化的生态系统中表征支撑微生物功能的关键蛋白质。作为微生物组的主要催化与结构组分,蛋白质能够提供关于微生物群落活性过程及生态功能的独特见解。通过将宏蛋白质组学与其他组学技术整合,研究人员能够全面解析微生物生态学特征、种间互作及功能动态。本综述由宏蛋白质组学倡议(Metaproteomics Initiative, www.metaproteomics.org)撰写,旨在为微生物组学和蛋白质组学研究人员提供实践指南,介绍了宏蛋白质组学所必需的关键原理、系统阐述宏蛋白质组学的核心原理、前沿技术及关键分析流程,涵盖实验设计、样本制备、质谱技术、数据分析策略与统计方法等全链条研究要素。
视频解读
Bilibili:https://www.bilibili.com/video/BV1h1VbzHEiK/
Youtube:https://youtu.be/2hYp1LCwe1k
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
1. 引 言
在生物圈内几乎所有过程中,微生物组的重要性已日益凸显。由细菌、噬菌体、古菌、酵母、真菌、原生生物及病毒等组成的微生物群落,其物种组成呈现高度多样性特征。微生物组与其功能活动场域(包含基因、转录本、蛋白质及代谢物等分子元件)共同构成了一个完整的微生物系统。在多数情况下,微生物组在群落结构和功能网络层面均展现高度组织化特性,这凸显了系统解析微生物组与其环境(或真核宿主)间复杂互作关系的必要性——无论这些互作呈现共生还是致病特性。然而,这些系统的复杂性对传统研究工具提出了挑战,尤其是依赖培养的方法。鉴于微生物组内存在密集的种间互作网络,基于培养的研究范式难以扩展应用于大规模微生物组研究。
组学技术的快速发展为研究复杂微生物组的系统生物学开辟了新路径。其中,鸟枪法宏基因组学已被证实其相较于16S rRNA基因扩增子测序等传统技术具有显著优势。宏基因组学技术能帮助发现未被分离培养微生物物种的完整基因组信息,从而揭示微生物组的代谢和生理潜能。然而,该技术仅限于功能预测,无法直接表征实时生物学过程。为突破这一局限,宏转录组学、宏蛋白质组学及代谢组学等技术可提供特定环境条件下的基因表达与功能蛋白方面的重要见解。这些技术的协同应用有效弥合了从分类组成到基因组功能潜力,再到动态的、情境依赖性的蛋白功能响应的研究桥梁。
宏蛋白质组学能够全面解析微生物组中表达的蛋白质及其功能,量化它们的丰度,并表征其修饰、相互作用和亚细胞定位(图1)。蛋白质作为微生物组中主要的催化单元和结构元件,使得宏蛋白质组学能直接反映微生物组的表型特征。该技术不仅能提供功能通路的精细描述,还能检测与微生物结构重塑、稳态维持和酶活性调控相关的特定蛋白质变化。通过分析蛋白质序列的差异特征,研究人员能追溯特定功能酶系的物种来源,从而实现功能与分类单元的精准关联。
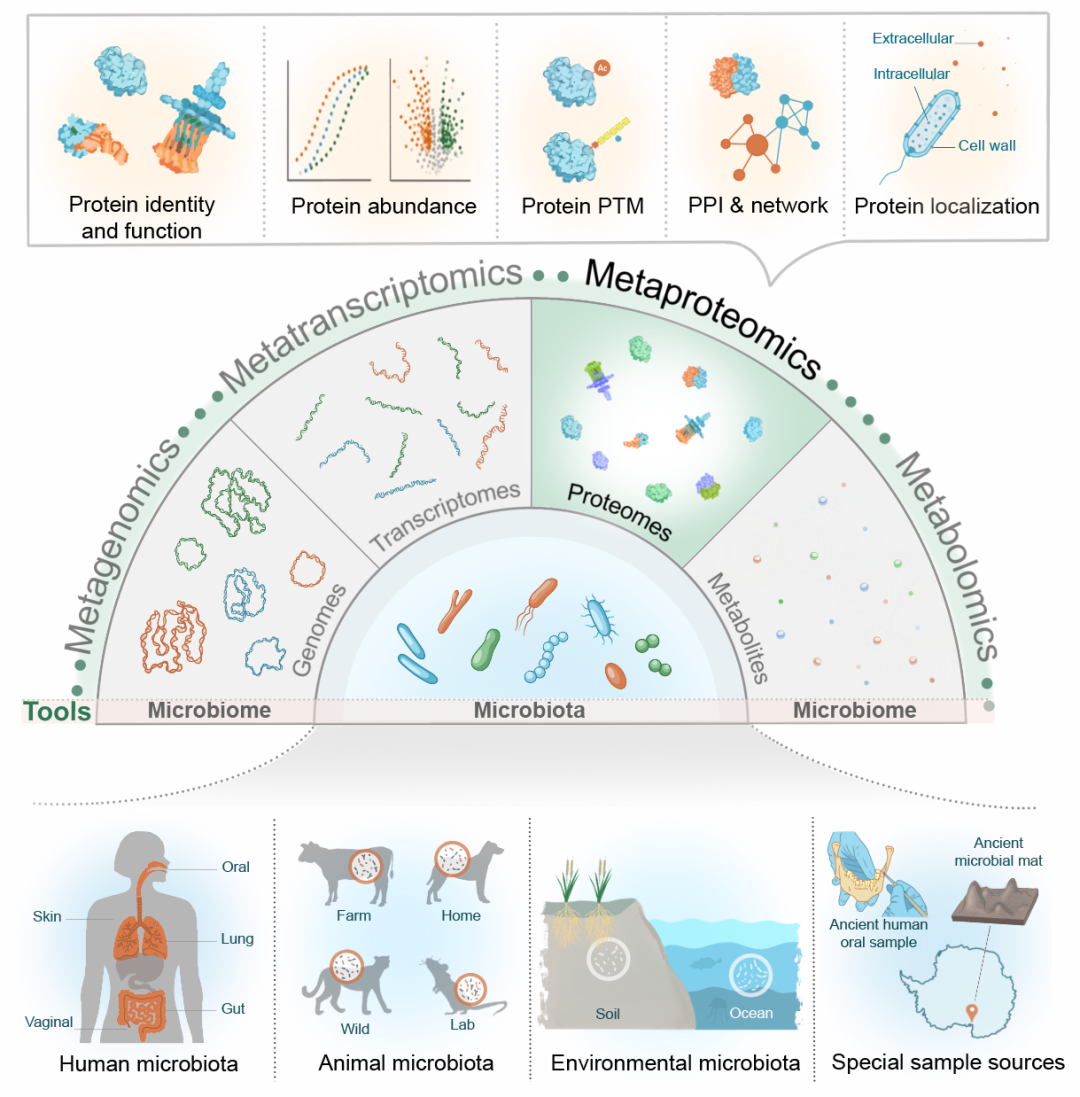
图1. 多重组学技术中宏蛋白质组学在不同微生物组研究领域的应用
本图展示了宏蛋白质组学在蛋白质鉴定、定量、PTMs检测、PPIs以及蛋白亚细胞定位中的应用。宏蛋白质组学可与宏基因组学、宏转录组学及代谢组学等其他组学方法互为补充,从而全面解析微生物组系统。微生物组示例研究领域包括人体微生物组(口腔、皮肤、肠道、肺部和阴道)、动物微生物组(家畜、野生动物和实验动物)、环境微生物组(土壤和海洋)及特殊来源的微生物组样本(如古代微生物组样本)。
宏蛋白质组学已在许多具有深远影响的研究中成功应用:在基础研究层面,深化了微生物生态学理论、宿主-微生物互作机制及病理机制的认知;在应用领域,优化了厌氧消化、废水处理等生物技术过程,提升了环境监测效能与农业生产效率;在交叉学科中,更拓展至历史文化遗产解析与法医物证鉴定等创新方向。关于本技术的系统评述及前沿展望,建议参阅相关专题论述。
我们接下来例举宏蛋白质组学技术的三个典型应用。首先是解析海洋中微生物活性以用于生物监测。海洋在全球气候调节、碳储存及环境污染治理中具有关键作用。理解稳态与动态环境条件下水生系统的碳氮固定过程及污染物降解机制,对完善气候变化监测和污染防控体系至关重要。近期宏蛋白质组学研究揭示了微量营养素在调控铵代谢与碳循环中的作用,并成功解析了跨尺度的生物地球化学过程。尤为重要的是,该技术实现了不同海域及深度的硝化作用与碳代谢过程图谱构建,并发现锌限制(通过锌响应蛋白表征)是调控海洋微生物与藻类活性的关键因素。同时,宏蛋白质组学阐明了深海环境中浮游动物、细菌、古菌及病毒在碳循环中的差异化作用。研究发现,深海中γ-变形菌虽丰度较低,但胞外酶的高表达(可能由噬菌体诱导的细胞裂解所致)能有效促进静水压力下的碳循环进程。总之,这些发现对海洋活动监测具有重要启示,可能使微生物清除蛋白能够用于生物监测,如追踪局部水域的锌水平。
第二个应用则是侧重于利用宏蛋白质组学技术优化生物燃料生产与饲料利用效率。在农产品(蔬菜、肉类)及生物燃料需求激增,以及塑料、废水等废弃物持续增长的背景下,提升生产效能与废弃物管理策略迫在眉睫。宏蛋白质组学有助于破译海洋微生物降解塑料的代谢通路,鉴定出由稀有类群合成的聚酰胺酶、水解酶及解聚酶,有望为工业级塑料降解提供了酶资源库。同样,利用宏蛋白质组学技术阐明木质纤维素生物燃料生产中的功能微生物及特异性碳水化合物活性酶,使得高固体含量条件下的工艺优化成为可能,这对工业化生产效率提升具有决定性意义。此外,微生物群落组成和碳水化合物活性酶在牛饲料利用效率中也发挥着重要作用。然而,仅有一篇近期基于瘤胃微生物组的宏蛋白质组学研究表明,蛋白质层面的功能冗余与生态位分化是影响饲料转化率的核心机制,这些发现为开发益生元/益生菌干预策略提供了理论基础,以应对资源约束与需求增长的双重挑战。
第三个应用聚焦于识别生物医学疾病标志物。人类微生物组因其在疾病发生、进展和治疗抵抗中的作用而受到广泛关注。尽管已针对多种疾病状态表征了菌群失调的群落特征,但不同微生物群落模式之间的功能冗余限制了其诊断应用价值,因此需要借助其他组学技术。近期,宏蛋白质组学技术深入揭示了炎症性肠病(Inflammatory bowel disease, IBD)患者中饮食、宿主与微生物组之间的复杂相互作用。该研究发现了新的生物标志物,这些标志物在炎症指标方面可能优于钙卫蛋白。值得注意的是,这些标志物在物种分类学层面是无法被检测的。该研究还发现粪便中的膳食蛋白、小肠吸收不良与炎症之间存在关联。这些发现凸显了宏蛋白质组学技术在研究饮食相关疾病和膳食干预方面的优势,因其可以独特地实现对微生物组组成、宿主响应和膳食成分的同步分析。
此外,宏蛋白质组学在这些应用领域的优势还体现在其能解答诸多重要问题:(i)微生物在多样化生境(包括环境、技术和宿主相关系统)中的代谢和生理过程是什么?(ii)微生物组如何通过表达差异蛋白来响应环境变化?(iii)微生物如何与胞外/胞内蛋白质动态所反映的微环境相互作用?(iv)哪些PTMs调控蛋白质活性和结构?(v)微生物组表型如何随时间或空间尺度发生变化?(vi)如何通过宏蛋白质组的稳定同位素信息表征微生物活性和底物利用?
本综述由宏蛋白质组学倡议(Metaproteomics Initiative)编写,旨在为宏蛋白质组学研究提供实用且易于掌握的指南。第5部分详细阐述了这项合作文章的组织过程与呈现形式,体现了我们为微生物组研究领域提供系统性、权威性资源的初心与集体努力。
2. 蛋白质组学基础
蛋白质是执行DNA遗传信息的关键结构与功能单位,广泛参与生命活动,从催化生化反应到构建细胞结构,几乎无所不包,构成了生命体系正常运作的基础。“蛋白质组(Proteomics)”这一术语指代细胞、组织或生物体中表达的全部蛋白质集合。蛋白质组学作为一门学科,致力于揭示蛋白质的身份、丰度、结构、相互作用及修饰特征,从而深入解析其在生物系统中的功能。
尽管“蛋白质组”的概念形成于20世纪90年代中期,但其基础源于几十年来的蛋白质生物化学研究,这些研究持续影响着现代蛋白质组学的发展。蛋白质组学最早的应用形式结合了凝胶电泳技术(一维与二维)与基质辅助激光解吸电离(Matrix-assisted laser desorption-ionization, MALDI)、电喷雾电离-液相色谱-串联质谱(Electrospray ionization-liquid chromatography-tandem mass spectrometry, ESI-LC-MS/MS)等质谱技术。在初期实验流程中,蛋白质样品通过一维/二维凝胶联合分离:一张凝胶经电转印至硝酸纤维素膜后利用氨基黑染色,另一张凝胶则使用灵敏度更高的银染法显色。研究者通过比对硝酸纤维素膜与银染凝胶的对应区域,可定位低灵敏度染色下难以识别的蛋白条带。随后从硝酸纤维素膜切取目标条带或斑点,经胰蛋白酶酶解后通过质谱进行鉴定。随着原位酶解技术的改进,电转印步骤被逐步淘汰。早期研究还催生了自动化蛋白质鉴定软件工具,替代了手工标注肽段序列的传统方法。这些早期的创新性成果为现代蛋白质组学工作流程奠定了基础。
基于非凝胶蛋白质组学的发展标志着该领域的重大突破。该方法跳过凝胶分离步骤,直接在蛋白质提取步骤后进行酶解与质谱分析。基于非凝胶蛋白质组学技术催生了一系列新技术、试剂(例如:稳定同位素氨基酸标记(Stable isotope labeling by amino acids in cell culture, SILAC)、同位素编码亲和标签(Isotope-coded affinity tags, ICAT)、等重标签相对与绝对定量(Isobaric tags for relative and absolute quantification, iTRAQ))和软件,这些共同提升了蛋白质鉴定、PTM分析、定量与多重检测能力。过去,使用二维凝胶电泳结合质谱技术(Mass spectrometry, MS)分析蛋白质需要经过繁琐的操作流程,然而,通过引入非凝胶工作流程使得分析更为高效便捷。此外,最初针对小分子研究优化的质谱仪经过改进后也应用于蛋白质组学。过去15年间,专为蛋白质组学设计的质谱仪逐步发展成熟,在肽段鉴定与定量方面展现出更高的速度、灵敏度与准确性。
当前,蛋白质组学方法主要分为两大类:鸟枪法(自下而上)蛋白质组学(Bottom-up proteomics)与自上而下的蛋白质组学(Top-down proteomics)。其中应用更广泛的是鸟枪法蛋白质组学,该方法通过酶解将蛋白质分解为肽段后进行质谱分析,在蛋白质鉴定与定量方面具有稳健性与高效性。而自上而下法则直接分析完整蛋白质,可解析其序列、结构及修饰特征。尽管自上而下法具有独特优势,但由于技术难度较高,在单一物种蛋白质组学中应用较少,并且尚未扩展至宏蛋白质组学研究。
典型的自下而上蛋白质组学工作流程始于蛋白质酶解(通常使用胰蛋白酶)生成短肽段,随后通过液相色谱分离并结合串联质谱(Liquid chromatography-tandem mass spectrometry, LC-MS/MS)进行分析。在质谱仪中,肽段经电离后首先检测其完整分子量生成MS1谱图,随后进一步碎裂产生MS2谱图。多数情况下,蛋白质组学搜库软件通过将实验谱图与蛋白质数据库的理论谱图进行匹配,实现肽段及其对应蛋白质的鉴定与定量。若需深入了解蛋白质组学技术原理,可参阅该领域详述文献资源。
3. 宏蛋白质组学的实验方法
宏蛋白质组学在蛋白质组学技术基础上拓展而来,通过利用高分辨率LC-MS/MS仪器及配套的质谱数据解析软件进行肽段鉴定。然而,宏蛋白质组学并非简单地将蛋白质组学方法应用于微生物组研究,其复杂性体现在需同时考虑每个蛋白质的物种特异性注释与功能注释。此外,样本中系统发育相关物种间存在的蛋白同源序列进一步增加了蛋白质推断的难度。
与传统蛋白质组学相比,宏蛋白质组学的核心差异在于:微生物组的物种分类与功能复杂性、庞大的微生物蛋白质序列数据库、样本前处理相关的挑战,以及肽段/蛋白质鉴定与定量的特殊性。同时,需要专门开发的生物信息学与统计学工具来追踪肽段及蛋白质的分类学与功能注释。这些独特的技术特征将在本文后续章节详细阐述。
本节为开展宏蛋白质组学研究提供基础性框架指南(图2)。我们将从实验设计(第3.1节)开始,逐步阐述样本采集、保存与预处理(第3.2-3.3节)、蛋白质样本制备:涵盖手动工作流程(第3.4-3.5节)和自动工作流程(第3.6节)以及质谱数据采集原理(第3.7节),最终深入解析宏蛋白质组学生物信息学分析流程(第4.1-4.3节)。
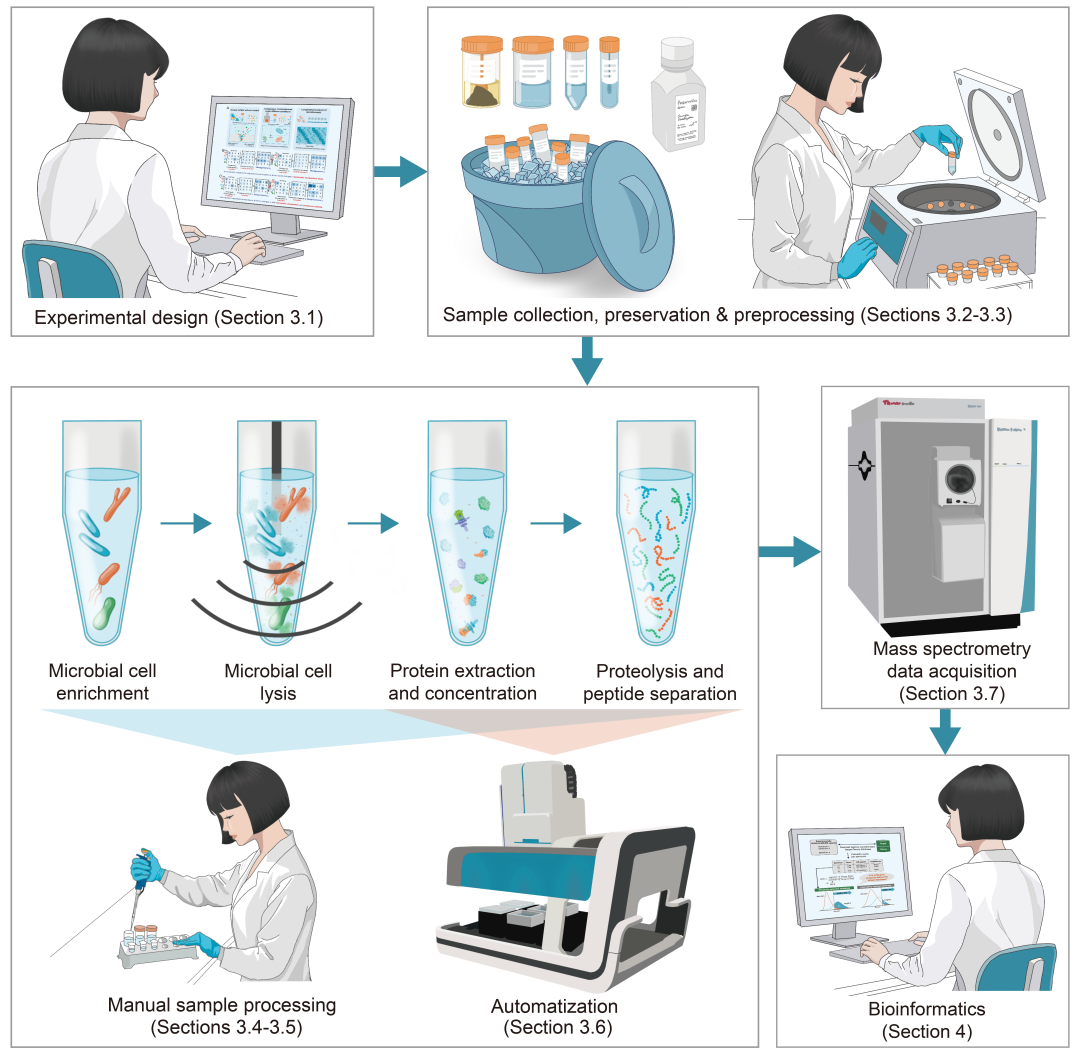
图2. 宏蛋白质组学技术的原理及实验工作流程概览
宏蛋白质组学工作流程一般从实验设计开始(第3.1节),随后进行样本收集、保存与预处理(第3.2-3.3节)。微生物细胞经过富集、裂解、蛋白质提取及肽段分离后,以上步骤可通过手动(第3.4-3.5节)或自动化(第3.6节)方式进行处理,随后进行质谱数据采集阶段(第3.7节)。最终通过生物信息学分析(第4节)进行数据库检索与数据解读,揭示微生物功能与生态学机制。
3.1 实验设计
3.1.1 实验设计与科学问题的适配性
一个设计合理的宏蛋白质组学实验是获得有价值科学见解的前提,必须紧密围绕所要回答的科学问题展开。尤为关键的是,实验设计应充分考虑研究目标的具体需求,并结合实际可用的样本类型、技术平台和数据分析资源,做到目标明确、策略得当、资源匹配。
总体上可概括为三类实验场景(图3A)。(1)无对照的单一样品分析:目标是对样本中存在的分类单元与功能单元进行全面描述,尽管无法进行对照比较。例如,利用宏蛋白质组学分析了南极历史冰芯样本的脱水物质、工业储液池的特殊生物膜、古墓残留物、中世纪牙结石。通过分析样本中不同微生物的功能丰度差异,可揭示其代谢特征。(2)不同条件下微生物组的比较分析:此常见策略聚焦于条件间的差异比较,可设计为两组(条件A对比条件B)或多组复杂实验。具体场景包括:剂量效应分析:改变单一参数(如应激强度)或空间梯度的比较。例如:沿着太平洋5000公里断面对微生物群落进行特征分析,或分析微生物组对外源性刺激的响应机制。(3)单一/多微生物组的纵向分析:通过时间序列采样捕捉微生物群落的动态变化(可能包含宿主响应)。复杂设计可结合多条件或多位点进行时序比较,例如:监测克罗恩病患者术后一年内的肠道微生物组变化,或用于木质纤维素分解的两段式厌氧消化系统中,对水解与产甲烷亚系统进行动态监测。
部分读者可能已具备设计宏基因组学实验的经验,并理解其基本原理。相比之下,宏蛋白质组学从另一个维度揭示微生物组的动态变化(图3B-C)。宏基因组学主要通过捕捉由物种组成变化引发的群落差异来反映样本之间的差异,这是基于样本内的基因组总量相对稳定(图3B)。因此,宏基因组学擅长揭示物种丰度与多样性,但对功能层面响应的洞察则较为有限。而宏蛋白质组学不仅可以利用分类特异性肽段的丰度信息来衡量物种组成的变化,还能够通过不同分类单元中蛋白质组的表达差异,捕捉微生物群落在功能上的动态响应(图3C)。正因如此,宏蛋白质组学在研究不同条件下微生物组的功能差异,或进行纵向时间序列研究时,具有独特的优势和应用价值。
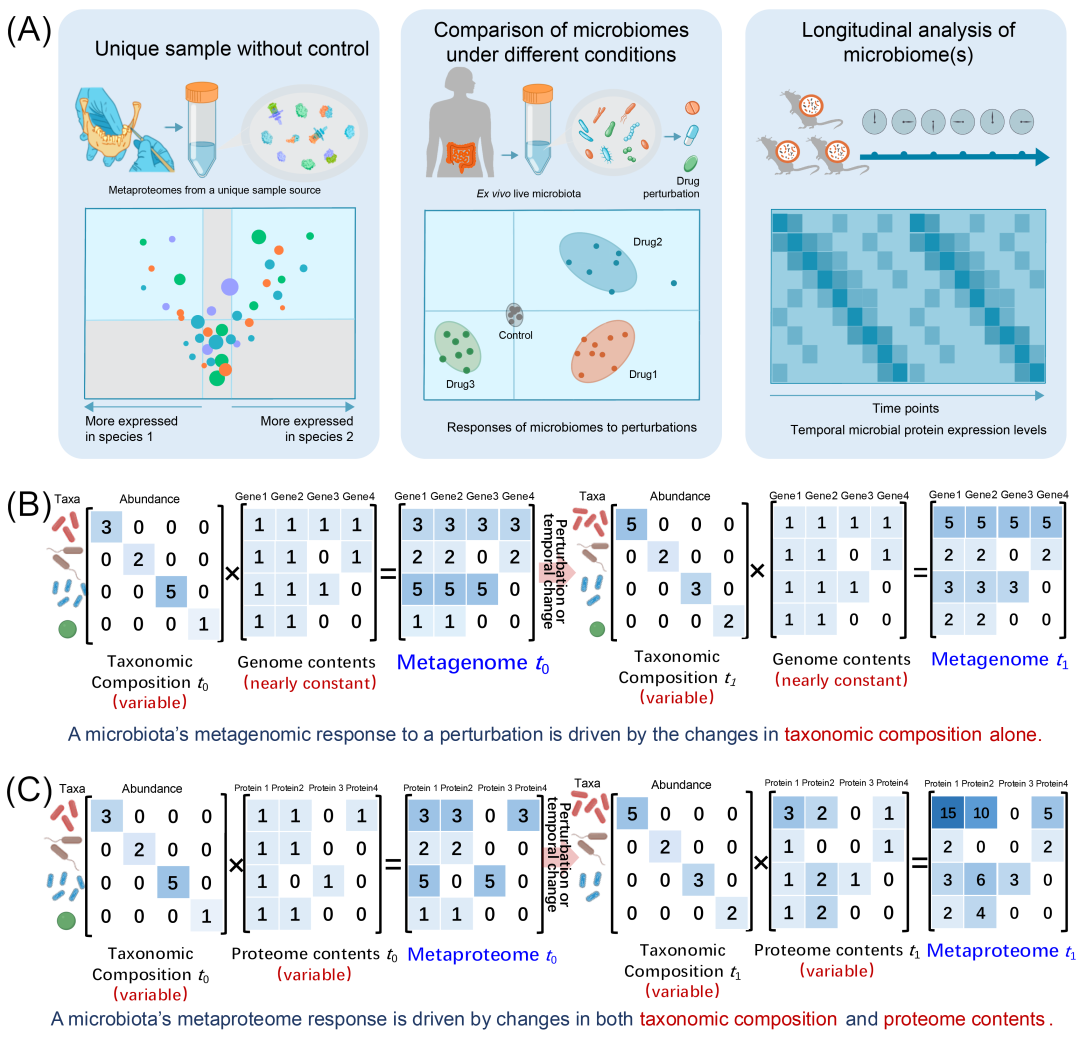
图3. 微生物组动态研究中宏蛋白质组学实验设计与宏基因组学的比较
(A)常见宏蛋白质组学实验设计概览。左图展示,在单一样本来源物种间蛋白质表达丰度比较(无对照组)。中图展示,体外培养微生物组,对外源性干扰(如药物处理)的微生物组功能响应分析。右图展示,纵向研究模式下监测微生物蛋白表达的时序变化。(B)宏基因组学对扰动的响应主要表现为物种组成变化,同时假设基因组内容保持相对稳定。(C)宏蛋白质组学扰动的响应,则表现为物种组成和蛋白质组表达的变化,可用于解析微生物丰度及其功能贡献的协同响应。
在为动力学分析选择条件或时间节点时,必须审慎考量。差异过大的样本(如土壤微生物组与人类肠道微生物组)的比较通常缺乏理论价值,而过于相似的样本可能不会显示出显著差异。样本的选择应基于明确的理论依据和前期观察结果。参照条件或时间点的设定取决于科学问题,但可能采用将所有样本混合作为参考的方式。虽然这种方法能增加参照样本中肽段的多样性,但如果分析流程无法全面覆盖样本的多样性(详见3.5与3.7节),可能会使分析过程变得复杂。
在实验设计阶段,还必须考虑潜在的混杂因素。因此,样本收集时,全面的元数据采集十分重要,包括采样位置、时间、存储条件、处理过程和数据来源等信息。其他元数据,如采样当天的天气、患者用药史或健康状况,也可能对解读结果产生重要影响。此外,研究人员还应考虑使用额外的样本来构建蛋白序列数据库,以便用于肽段谱图匹配,并在处理所有样本之前进行方法学验证。关于蛋白质组学软件及蛋白序列数据库构建的详细信息,请参见第4.1.1节和第4.1.2节。
最后,尽管少数宏蛋白质组学研究已经应用了代谢标记技术,但该方法通常对环境或人体微生物组样本缺乏可行性。代谢标记技术,如第2节中简要提到的,涉及通过将15N或13C等重同位素整合至蛋白质进行底物标记,从而用于研究代谢互作与蛋白质合成速率。然而,鉴于其适用性有限,本综述不再深入讨论该技术。
3.1.2 数据可重复性与统计学分析
宏蛋白质组学样本具有高度复杂性和异质性特点,因此在实验设计过程中需充分考虑统计学功效,确保结果具有可重复性。生物学重复、技术重复和分析重复是获得可靠数据与准确解读结果的关键。增加生物学重复的数量有助于在高变异性的背景下提高对微小差异的检测能力。当实验条件之间只存在微小差异时,也可考虑使用合并样本。技术重复和分析重复对于消除测量过程中引入的噪声至关重要。建议首先使用代表性样本评估样本制备及分析流程的变异性。此外,在LC-MS/MS分析前随机化样本顺序,有助于降低由于分析顺序导致的偏倚风险。对于已知存在批次效应等特定变异来源的情况,推荐采用分组随机化方法以进一步控制偏倚。严格的质量控制(Quality control, QC)程序在 LC-MS/MS 分析阶段至关重要,以确保数据的可靠性和一致性。关于质控措施详见第3.7.4节。
确定合适的生物学重复数量对于检测生物学意义的差异(如物种生物量、蛋白质丰度或代谢通路的变化)至关重要。统计学功效分析通常用于计算所需的样本量,但由于宏蛋白质组学实验设计的复杂性和样本固有的高度变异性,该方法的应用可能存在挑战。当缺乏精确终点参数时,可参考同类研究的经验值进行估算。功效分析需综合考虑以下关键参数:效应量,它反映了组间差异的预期大小,并帮助确定所需样本量;显著性水平(α),通常设置为0.05,允许5%的假阳性风险;统计功效(1 - β),通常设定为0.8或更高,以降低假阴性概率;数据变异性,可通过预实验或同类文献数据估算。针对复杂微生物群落的研究,虽难以获得精确样本量估值,但近似估算仍具有重要参考价值。开展功效分析既能避免统计效力不足的研究设计,又可实现资源配置的优化。
3.2 预处理前样本的采集、保存、和储存
3.2.1 样本采集和保存
宏蛋白质组学已广泛应用于多种样本类型的研究,包括水体、土壤、污水、气溶胶及岩石等环境微生物群落,以及发酵食品和饮料中的微生物组。此外,宏蛋白质组学还用于研究多种高等真核生物的共生微生物群,包括蛛形纲、昆虫、蠕虫、软体动物、鱼类、植物、鸟类和哺乳动物。在哺乳动物及其他脊椎动物中,该技术已被应用于分析消化、呼吸及泌尿生殖系统等不同部位的微生物组特征。然而,仍有大量微生物群落尚未进行宏蛋白质组学研究。
研究人员通常直接采集微生物组样本至无菌容器中。对于非侵入性的临床样本(如粪便、唾液、痰液和尿液),通常由受试者自行完成采样。对于需要表面取样的临床标本,如口腔、鼻腔及宫颈阴道样本,常使用拭子、刮勺或注射器进行采集;而针对牙齿及牙龈相关微生物群,常采用牙周刮匙或滤纸条。部分样本集需借助侵入性手段,如支气管肺泡灌洗、气管内吸引、肠道活检、结肠腔抽吸或通过手术获取结肠内容物。此外,在实验动物或野生动物研究中,可利用胃肠瘘管技术或尸检解剖等方式收集样本。对于环境样本,常采用专业设备,如用于生物溶胶的石英滤膜收集器,以及用于水体样本的大体积转移/过滤系统。复杂生态系统样本可能需要多步骤采集方案。
采样方法的选择对样本的宏蛋白质组分析结果具有重要影响,包括微生物与非微生物成分的比例以及微生物不同分类群的相对丰度。此外,样本采集策略会引入操作人员相关的人为差异,因此,对于临床样本的自采集而言,操作简便的设备更具优势,因其可提高可重复性。为优化采样方法,目前亟需开展方法学研究以明确:不同采样方案对宏蛋白质组学结果的潜在影响,同一环境或宿主相关微生物群落不同样本类型间的可比性。此外,在样本采集与样本处理过程中,还需特别注意避免常见污染,例如来自塑料器皿的聚乙二醇(Polyethylene glycol, PEG)污染,以及由于人工操作引入的角蛋白污染。
微生物组采样本质上是一个将微生物群落从其原始生态环境转移至实验室条件的过程。在此过程中,微生物群对外界环境变化(如温度、湿度及化学或生物因子暴露)极为敏感,这些因素会显著影响宏蛋白质组的组成。为尽可能减少干扰因素,理想情况下应在采样后立即进行蛋白质提取。然而,在大规模研究或野外采样中,样本往往难以立刻处理。因此,合理的运输和储存措施对于维持微生物群的生物学特征至关重要。特别是对于低生物量或低多样性的微生物群,其组成和活性更易因外界刺激而迅速变化,因此,妥善的保存策略对于确保宏蛋白质组数据的可靠性和生物学代表性至关重要。
3.2.2 维持样本完整性的储存条件
规范的存储条件对于维持微生物蛋白完整性并确保后续分析可靠性具有决定性作用。样本暴露于变化的环境中(如接触空气、温度波动或营养耗竭),蛋白质组特征可能发生显著变化,从而导致误导性结果。例如,空气暴露可能引发氧化应激,进而导致细菌超氧化物歧化酶的富集,这在结直肠癌研究中可能产生偏倚,使其误认为是疾病特异性特征。因此,在采样后应立即采取适当的存储措施,以最大程度地保持微生物群的原始状态。
标准的宏蛋白质组学样本保存方法包括液氮速冻后存储于-80 °C长期存储,以最大限度地减少分子降解并维持蛋白丰度的稳定性。尽管该方法十分有效,但在某些实验条件下,可能难以实现即时冷冻。在此情况下,可采用替代保存方法,例如磷酸盐缓冲液(PBS)、Amies液体培养基、NAP缓冲液及其他商业化保存液,以优化存储条件或实现常温保存。此外,为防止生物体液(如唾液)中的蛋白水解,通常会添加蛋白酶抑制剂。RNAlater及类似试剂已被用于稳定肠道及海洋样本的蛋白质,尽管其效果仍存在争议。无论采用何种保存方法,都必须确保其与后续蛋白质提取、酶解及分析流程兼容。为系统评估不同保存方法的有效性和稳定性,国际多中心研究“宏蛋白质组学关键评估计划-2”(Critical Assessment of MetaProteome Investigation-2, CAMPI-2)针对各预处理步骤进行严格控制,以减少操作员的人为差异,从而识别潜在偏倚并验证方法的可重复性。
此外,还可采用其他长期存储策略,如冻干、−20 °C冷冻、液氮储存或制备冻干粉末等。然而,这些方法的有效性需针对不同样本类型进行严格评估和验证。这些策略可能仅适用于特定类型的样本。例如,研究表明,相较于直接存储蛋白质提取物,将完整的粪便样本冷冻于−80 °C具有更高的稳定性,这进一步凸显了根据样本特性选择合适存储策略的重要。
需要注意的是,蛋白质在存储过程中的稳定性高度依赖于样本类型及存储条件。例如,土壤蛋白的活性与稳定性受存储温度、持续时间及土壤有机质含量等多种因素的影响。对于涉及长时间运输或存储的研究,建议引入已明确组成的模拟微生物群落作为对照,以评估样本稳定性并检测的存储过程中可能引发的生物学变化。
3.3 样本预处理
样本预处理的主要目的是去除污染物和杂质,以避免影响蛋白质提取、降低后续分析质量,并稀释具有生物学意义的信号。与基因表达检测流程类似,合理的预处理有助于富集微生物组分,从而提升下游分析的质量。理想情况下,样本预处理应尽可能简化、快速且具备良好的重复性。然而,目前宏蛋白质组学(或宏基因组学)尚未建立统一的标准化流程,因此具体方法需根据样本类型进行优化,并结合研究目标加以评估。尽管目前已用于宏蛋白质组学分析的样本类型范围仍较有限,随着该领域的迅速发展,预计将有更多技术方法不断问世。
对于土壤样本,由分解的有机物质衍生的腐殖质常与蛋白质共同被提取,进而干扰质谱检测。为解决这一问题,研究者已开发多种方法,在酶解前去除腐殖质化合物的同时保持蛋白质的完整性。此外,滤膜辅助样本制备(Filter-aided sample preparation, FASP)方法可直接在腐殖质化合物中消化酶解蛋白质,该方法通过酸化沉淀腐殖质及未消化的蛋白质,并利用分子量截留滤膜进行离心,以获取目标肽段。
对于人肠道微生物组样本,宿主细胞及食物残渣中的非微生物蛋白通常远多于微生物蛋白,从而降低微生物宏蛋白质组鉴定效率。为提高鉴定率,可采用双级过滤和差速离心等方法富集微生物细胞。然而,这些方法可能引入选择性偏倚,需结合研究目标谨慎选择适用的富集策略。例如,双级过滤可去除宿主细胞及胞外蛋白,而差速离心可能导致特定微生物类群的非特异性损失。此外,这些方法通常较为耗时,并易受粪便样本个体差异(如质地、纤维含量及含水量)的影响。在大规模纵向研究中,推荐采用自动化技术(如固相萃取净化技术)以提升处理效率、降低操作误差,并提高数据的稳定性和可靠性。
在分析具有高度异质性的样本(如囊性纤维化患者的粘性痰液、某些植物组织或环境样本)的研究中,均质化处理有助于提高样本的一致性。此步骤应在严格控制温度和时间的条件下进行,以最大限度减少对样本宏蛋白质组的干扰。常见的机械均质化方法包括实验室研磨机和玻璃匀浆器。此外,操作过程中可添加蛋白酶抑制剂和DNase I,以防止蛋白降解并破坏DNA聚集物,但其适用性应根据样本类型和研究目标进行谨慎评估。
对于含有细菌或病毒病原体的临床样本,如需在相应生物安全防护等级(Biosafety level, BSL)之外进行后续处理,必须先进行灭活操作。由于目前尚无标准化的灭活流程,因此必须根据病原体类型和样本特性定制灭活方案。常用的灭活方法包括在锂十二烷基硫酸盐缓冲液中进行热灭活,以及代谢物–蛋白质–脂质联合提取(Metabolite, protein, and lipid extraction, MPLEx)法,该方法利用氯仿-甲醇-水(8:4:3)体系同时实现病原体灭活,并将样本分为代谢物、蛋白质和脂质组分。这些方法不仅能确保安全性,还能与后续的宏蛋白质组学分析流程兼容。
3.4 蛋白样本制备:从蛋白提取到蛋白酶解
从生物材料中制备蛋白质样本涉及一系列相互关联的关键步骤,每个步骤对获得高质量的宏蛋白质组学数据均至关重要。“蛋白质提取”这一术语常被广义用于描述从生物样本中分离蛋白质的整体流程。该过程通常以裂解缓冲液进行细胞裂解为起点,可能包括后续的蛋白纯化步骤,如蛋白沉淀、蛋白过滤等。部分实验方案将提取、纯化与酶解整合为单一流程,此时蛋白质纯化被视为一个独立的步骤。本节系统阐述蛋白质样本制备的核心环节:细胞裂解与蛋白提取(第3.4.1节)、蛋白纯化(第3.4.2节)、蛋白定量(第3.4.3节)和蛋白酶解(第3.4.4节)。
3.4.1 细胞裂解与蛋白质提取
细胞裂解可释放微生物细胞内的蛋白质组,目前已有多种裂解方法可供选择,每种方法均具有其特定优势。常见的机械破碎方法包括直接超声、非接触式超声及珠磨法。超声裂解通常分为直接超声(将超声探头直接插入样品中)和非接触式超声(样品置于离心管中,通过耦合液接收来自杯状超声头的超声能量)。其中自适应聚焦声波(Adaptive Focused Acoustic, AFA)技术作为一种先进的非接触式超声方法,可精确调控振幅、持续时间等参数,从而提高裂解效率,并最大程度减少蛋白质变性。珠磨法利用氧化锆或二氧化硅微珠实现细胞破碎,其中微珠粒径可影响破碎效率。
化学裂解方法主要依赖去垢剂,例如含Triton X-100或十二烷基硫酸钠(Sodium dodecyl sulfate, SDS)的尿素缓冲液,可有效破坏微生物细胞膜,并常与机械破碎或超声裂解联合使用。需要注意的是,当使用含尿素的缓冲液结合机械破碎或超声处理时,样品过热可能引发尿素诱导的氨甲酰化。此外,物理方法(如反复冻融或高压均质处理)同样可实现有效裂解细胞,其中压力参数需针对不同样品类型进行优化。由于微生物细胞结构存在显著差异,例如革兰氏阳性菌、革兰氏阴性菌和真菌的细胞结构不同,因此优化裂解条件对维持蛋白质完整性、提升蛋白得率以及确保无偏提取至关重要。
近年来,对上述部分方法的比较研究表明,在微生物组样本中,采用含尿素和SDS的裂解缓冲液结合超声裂解,相较于珠磨法,可获得更高的蛋白得率,并且样品本损失较少,尽管两者在肽段和蛋白鉴定数量上差异不大。在选择裂解缓冲液时,还需注意避免干扰后续质谱分析。例如,Tween-20等去垢剂可能会导致离子抑制,因此除非在后续清洗过程中去除(如利用悬浮捕获(Suspension trapping, S-trap)或FASP方法),否则应避免使用。
表1对比了常用的蛋白质样品制备方法,并总结了其主要优缺点。裂解方法的选择需综合考虑样本类型、目标蛋白的得率及其对变性或降解的敏感性。此外,不同裂解方法可以组合使用,例如,在复杂的微生物组样本中,含去垢剂的尿素裂解缓冲液常与超声处理联用,以实现微生物组样本中细菌的高效、无偏裂解。
表1. 标准蛋白样品制备方法的比较
本表总结了常见的蛋白样本制备技术,并概述其主要优势及潜在局限性。

3.4.2 蛋白纯化:沉淀和替代方法
蛋白质沉淀方法可通过去除脂类、核酸和多糖等可能干扰后续质谱分析的污染物,特别适用于复杂环境样本和粪便样本。在微生物细胞裂解后,从细胞碎片及污染物中高效分离蛋白质是确保高得率和高纯度的关键步骤。污染物的去除不仅能提高蛋白质得率,还能增强质谱检测的灵敏度,从而实现更精准、可靠的蛋白鉴定。
三氯乙酸(Trichloroacetic acid, TCA)/丙酮沉淀法是目前广泛应用的蛋白质沉淀方法。其原理是向蛋白裂解液中加入预冷(−20 ℃)的TCA或丙酮,或两者混合使用,以促进蛋白质变性沉淀,随后通过离心收集蛋白沉淀。随后,用冰丙酮(−20 ℃)清洗沉淀,以去除残留污染物及不溶性颗粒。该方法已被证实可在海洋沉积物、森林土壤等复杂基质样本中高效沉淀蛋白。此外,酸化的丙酮/乙醇缓冲液也被用于宏蛋白质组研究。
另一种常见的替代方法是苯酚萃取法。该方法利用有机相和水相的分配特性,使蛋白质进入有机相,而核酸则保留于水相,从而实现有效分离。苯酚萃取法尤其适用于富含有机和无机污染物的复杂样本,如土壤和废水污泥。该方法能够显著减少污染物干扰,从而提高目标蛋白的下游分析效果。此外,苯酚萃取法还能同步提取同一样本的核酸和蛋白质,使其在多组学整合研究(尤其是微生物组研究)中具有独特优势。
对于微生物含量较低的样本(如粪便样本、河床沉积物或空气过滤样本),最大化蛋白质得率至关重要。有机溶剂系统(如氯仿/甲醇或氯仿/甲醇/水混合体系)已被证明可通过优化溶剂比例和实验条件,提高蛋白质得率并减少低丰度蛋白的损失。此外,苯酚/氯仿双相体系或Triton X-114温度诱导相分离法能够选择性去除污染物,从而提高样本的纯度。
尽管传统蛋白沉淀方法有效,但其操作繁琐,且难以完全去除干扰物。为克服这些局限性,近年来开发了多种替代技术,以提高蛋白纯化及酶解效率。例如, FASP、单管固相增强法(Single-pot, solid-phase-enhanced sample preparation, SP3)和S-Trap等技术应运而生,这些方法将蛋白提取、纯化与酶解步骤整合为单一流程,显著提升了粪便等复杂样本的处理效率。此外,固相烷基化技术作为一种新兴方法,利用共价结合和蛋白质纯化策略,能够在降低样本损失和抗干扰条件下实现高效样品制备。该技术已成功应用于海洋微生物组样本,为高通量宏蛋白质组研究提供了新的思路。
3.4.3 蛋白质浓度测定
准确测定蛋白质浓度对于下游LC-MS/MS分析至关重要,可确保质谱分析时均一上样,提高数据获取的可靠性。在LC-MS/MS分析中,肽段的均一上样不仅直接影响定量准确性,还可保证不同样本间肽段的稳定检测。此外,均一上样有助于优化色谱柱性能,减少峰形畸变与保留时间波动,从而最大程度降低技术误差,提高样本间生物学差异的解析能力。
常见的蛋白质浓度测定方法包括比色法和荧光法。Bradford法基于考马斯亮蓝染料的比色原理,通过与蛋白质结合引起吸光度变化进行定量,并需使用已知浓度的标准品绘制标准曲线以确保准确性。双缩脲酸(Bicinchoninic acid, BCA)法则依赖于二价铜离子被蛋白质还原,并与双辛可宁酸形成紫色络合物进行定量,其灵敏度可通过调整试剂比例和孵育条件优化。荧光定量法(如Qubit蛋白质定量试剂盒)利用染料结合技术,可对低浓度蛋白样本进行高灵敏度定量,且受缓冲液成分干扰小。
此外,2-D Quant试剂盒通过选择性沉淀蛋白质,同时将干扰物质保留在溶液中,其显色深度与蛋白浓度呈负相关,线性检测范围为0–50 µg,适用于 1–50 µL体积的样本。实验应综合考虑目标灵敏度、动态范围及缓冲液兼容性。例如,SDS和苯甲基磺酰氟(Phenylmethylsulfonyl fluoride, PMSF)等蛋白酶抑制剂可能干扰部分检测体系,影响测定结果。
当无适配的蛋白定量方法时,可采用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(Sodium dodecyl-sulfate polyacrylamide gel electrophoresis, SDS-PAGE)粗略评估蛋白质浓度。尽管该方法精度较低,但在特定情况下仍可作为替代方案。通过比较目标条带与标准蛋白的染色强度,可快速判断样本间蛋白浓度的相对差异,为后续实验设计提供参考。
3.4.4 蛋白酶解
在自下而上(鸟枪法)的宏蛋白质组学研究中,蛋白质需要经过酶解处理,切割为肽段,以实现非靶向蛋白质鉴定。该过程包含多个预处理步骤,以确保高效酶解。首先,使用尿素或盐酸胍等变性剂促使蛋白质变性,从而暴露酶切位点。随后,采用二硫苏糖醇(Dithiothreitol, DTT)或三(2-羧乙基)膦(Tris (2-carboxyethyl) phosphine, TCEP)等还原剂断裂二硫键。为防止二硫键重新形成,需通过碘乙酰胺等烷基化试剂对半胱氨酸残基进行烷基化,使其硫氢基(-SH)转化为稳定的硫醚加合物。该烷基化过程会引入分子量变化,在肽段鉴定时需加以校正(详见4.1.1节)。
完成上述预处理后,蛋白质被酶切分解为适用于LC-MS/MS分析的肽段。胰蛋白酶是最常用的蛋白酶,因其高特异性和高效性,可在赖氨酸和精氨酸残基的C端切割蛋白质,从而生成适合质谱分析的肽段。此外,Lys-C蛋白酶可在高浓度尿素(8 M)条件下特异性切割赖氨酸C端,常与胰蛋白酶联用,以提升酶解效率和肽段覆盖度。其他蛋白酶如糜蛋白酶、Glu-C和Asp-N也可用于增加肽段多样性或满足特定研究需求。然而,胰蛋白酶与Lys-C的组合因其广泛适用性和高实用性,仍为最常见的选择。
酶与底物比例是影响酶解效率的重要因素,通常控制在1:50 至 1:100(w/w)。此外,酶解时间同样关键,一般在37°C条件下孵育数小时甚至过夜孵育,具体时间取决于样本的复杂性和酶的特性。酶解反应通常通过酸化终止,例如加入甲酸或三氟乙酸,将pH调至2–3。在某些方法(如S-trap或FASP)中,肽段可无需酸化直接洗脱。
消化后的肽段通常需脱盐或纯化,以去除盐分和杂质。常见方法包括固相萃取(Solid-phase extraction, SPE)、C18 ZipTips(Millipore)或超滤。在某些情况下,若液相色谱系统配备捕获柱进行在线脱盐,则该步骤可被省略。
近年来,溶液内酶解方法已被广泛应用,以简化实验流程并提高通量。代表性技术包括SP3、FASP、S-trap及基于in-StageTip(iST)的商业化试剂盒。这些方法可确保高蛋白得率,并与下游质谱分析高度兼容,尤其适用于微量样本的高效处理。
3.5 分离和分馏技术
分离与分馏技术能够降低样本复杂度,提高蛋白质鉴定与定量的深度和灵敏度。这些过程可在多个层级进行,包括肽段、蛋白质及细胞水平,具体取决于分析目标。例如,肽段分馏常用于增强LC-MS/MS的分析深度,而富集方法可实现特定PTM的靶向分析。在蛋白质或细胞水平,分馏策略可进一步降低样本复杂度或富集特定目标组分。
3.5.1 在线和离线肽段分馏
肽段分馏工作流程通常可分为一维(One-dimensional, 1D)、二维(Two-dimensional, 2D)或多维方法。在宏蛋白质组学研究中,一维液相色谱(1D-LC)是最常用的策略,其中反相(Reverse-phase, RP)纳升级高效液相色谱(nano-high-performance liquid chromatography, nanoHPLC,通常简称为LC或HPLC)利用C18色谱柱,根据肽段的疏水性进行分馏,并直接与质谱联用进行肽段分析。相比之下,二维液相色谱(2D-LC)通常基于多维蛋白质鉴定技术(Multidimensional protein identification technology, MudPIT),结合强阳离子交换(Strong cation exchange, SCX)与反相高效液相色谱(RP-HPLC)。肽段首先在SCX色谱柱上依据电荷进行分离,并通过盐梯度或pH梯度洗脱,随后在RP-HPLC色谱柱上进一步按照疏水性进行二次分离。2D-LC策略已成功应用于宏蛋白质组学研究,以提高鉴定深度,其中在线2D LC-MS联用系统已被用于人类肠道和环境微生物群的鸟枪法蛋白质组学研究。
尽管离线预分馏由于操作繁琐且需要增加质谱分析时间,在宏蛋白质组学研究中应用较少,但其在提高肽段和蛋白质鉴定深度方面具有较大潜力。高pH反相色谱法是一种常见的离线分馏方法,与低pH反相液相色谱-质谱梯度互补。此类分馏可通过层析柱尖端分步法(stage-tip)实现,也可通过高效液相色谱系统完成。其中层析柱尖端分步法操作简便,且已有市售试剂盒(如Pierce™高pH反相肽段分馏试剂盒)可供使用。另一方面,微流控高效液相色谱系统则可通过连续收集大量馏分并进行阶梯式级联,可实现更高分辨率的肽段分馏。
虽然深度分馏可显著提高宏蛋白质组学分析的深度,但同时也增加了实验成本、样本需求量及质谱分析时间,因此在大规模队列研究中应用受限。多重标记技术(如串联质谱标签,(Tandem mass tags, TMT))的应用可有效缓解这些限制,该策略减少了质谱分析时间,并降低了单个实验条件所需的样本量。离线肽段分馏与多重标记技术的结合,为研究人员(尤其是初学者)提供了一种可行的解决方案,使其能够开展深入的宏蛋白质组学研究,以探索微生物群的功能特性。
3.5.2 翻译后修饰肽的富集
翻译后修饰(PTMs)是蛋白质活性与功能的关键调控因子,而宏蛋白质组学技术在此类研究中具有独特优势。与其他组学方法不同,宏蛋白质组学能够直接鉴定和定量微生物蛋白中的PTMs,从而揭示微生物群的功能特性。尽管在宏蛋白质组水平分析PTMs极具挑战性,但已有多项研究成功在环境和人类肠道微生物组开展了metaPTMomics研究。这些研究鉴定了甲基化、羟基化、酰基化、瓜氨酸化、脱氨化、磷酸化及亚硝基化等多种PTMs,并发现其丰度在不同类型的微生物群中存在显著差异。理解PTMs的多样性及其分布规律对于揭示微生物群的功能至关重要。近年来,该领域的最新进展已在两篇综述中得到了系统阐述。
微生物组PTMs的研究可基于非富集样本,结合特定的生物信息学分析流程,也可通过肽段或蛋白水平的富集技术进行定量分析。根据PTMs的类型,可采用针对性的富集策略,以提高质谱检测的灵敏度。
免疫亲和富集技术广泛用于赖氨酸乙酰化、丙酰化和丁二酸化等蛋白质酰基化的研究,并已成功应用于人类肠道微生物组。该技术利用偶联至琼脂糖或磁珠的抗体,选择性富集酰基化肽段,从而显著提升质谱检测的灵敏度和特异性。然而,该方法受限于基序特异性抗体的可及性,且难以覆盖所有修饰类型的肽段。
固定化金属亲和色谱(Immobilized metal affinity chromatography, IMAC)是蛋白质组学中用于富集磷酸化肽段的常用策略,在磷酸化蛋白质组学研究中得到广泛应用。典型代表包括钛离子IMAC(Ti-IMAC)和铁离子IMAC(Fe-IMAC),可在LC-MS/MS分析前实现高效富集。
亲水作用液相色谱(Hydrophilic interaction liquid chromatography, HILIC)是另一种有效的富集策略,尤其适用于糖肽的富集。该方法依赖其对亲水性聚糖结构域的高度选择性和特异性,实现精准分离。这些富集技术已广泛应用于哺乳动物细胞、组织及单一菌株研究,并在微生物组研究中展现出广阔的应用潜力。
3.5.3 蛋白质、细胞水平及功能分馏技术
微生物群的高度复杂性通常需要在肽段分级的基础上,进一步进行细胞和蛋白质水平的分离,从而提升宏蛋白质组分析的深度和分辨率。尽管高通量、高分辨率的质谱仪的出现使得肽段分馏足以满足多数蛋白质组学工作流程的需求,但由于微生物群的复杂性,上游分级方法仍然具有重要价值。
毛细管区带电泳(Capillary zone electrophoresis, CZE)是一种用于分离带电分子的技术,在完整蛋白甚至细菌细胞的分离方面展现出应用潜力。另一种基于细胞壁结构差异的细菌分型方法是差异裂解法。尽管该方法的分辨率相对较低,但它可以根据细胞壁结构的差异区分不同类型的细菌。该方法通过使用梯度增强型裂解缓冲液(如含有尿素或不同浓度SDS的缓冲液)实现分步裂解,从而将细胞壁较薄的革兰氏阴性菌与具有多层厚壁结构的革兰氏阳性菌的蛋白质组进行分离。
在宿主相关的微生物组研究中,为增强微生物信号的检测,去除高丰度宿主细胞通常至关重要。差速离心与密度梯度离心是常用于富集微生物细胞的技术。在微生物细胞裂解后,还可通过超速离心等方法进一步分离细胞组分,从而提高蛋白质鉴定的覆盖率。
功能性分馏技术,如基于活性的蛋白质探针技术(Activity-Based Protein Probing, ABPP),可用于在蛋白质组水平研究酶功能。ABPP采用小分子探针共价结合特定功能或特定残基的蛋白活性位点,随后对这些标记蛋白进行富集或捕获,并通过LC-MS/MS进行分析,从而实现蛋白功能的精细解析,并有助于药物靶点的发现。该技术在解析未知功能蛋白方面尤其具有应用价值,因此在微生物组研究中具有重要意义。近年来,ABPP在宿主相关及环境微生物组研究中的应用,揭示了多种微生物酶的功能,包括含巯基蛋白酶、胆盐水解酶(Bile salt hydrolases, BSHs)、糖苷水解酶(Glycoside hydrolases, GHs)以及β-葡萄糖醛酸苷酶等。
3.6 自动化技术
高通量技术的应用实现了样本制备流程的革新,通过简化繁琐的人工操作步骤,彻底改变了蛋白质组学的工作流程,随着数据集规模和复杂性的持续增长,这一转变显得尤为重要。这些技术进步不仅推动了化学蛋白质组学、生物标志物检测以及药物靶点发现等领域的应用。尽管宏蛋白质组学的自动化技术发展速度不如蛋白质组学迅速,但其推动领域变革的潜力依然巨大。
自动化宏蛋白质组学工作流程具有多项优势,包括缩短样本处理时间、减少人为误差并提高实验的可重复性。这些改进使研究人员能够在有限的实验周期内,更全面地解析微生物组对环境因子的响应。此外,高通量自动化流程使得研究人员能够大规模发掘与微生物组相关的生物标志物,并深入探索不同微生物组的动态功能景观。自动化技术还能够生成大规模数据集,可为人工智能(Artificial intelligence, AI)技术提供分析基础,从而揭示宏蛋白质组学数据中隐藏的模式与规律。
宏蛋白质组学的自动化样本处理流程通常包括四个关键步骤:微生物细胞裂解与蛋白质提取(第3.6.1节)、蛋白酶解与肽段纯化(第3.6.2节)以及多重分析(第3.6.3节)。
3.6.1 微生物细胞裂解与蛋白质提取
在处理复杂临床样本(如人类粪便或唾液)时,通常需要进行微生物细胞富集,但这也面临显著挑战。数据集中样本的特性可能存在很大差异,这使得自动化微生物细胞纯化的技术参数标准化变得更加复杂。因此,目前的自动化宏蛋白质组学工作流程通常不包括全自动原始样本处理步骤。例如,RapidAIM 2.0工作流程需结合手动微生物细胞富集和细胞清洗步骤,同时使用96通道液体处理工作站加速移液操作。相比之下,SHT-Pro实验步骤,作为首个专为大规模粪便样本处理设计的高通量流程,直接从原始粪便样本裂解开始,无需预先进行微生物富集。这种方法特别适用于需要同时研究宿主和微生物蛋白质的情况。
微生物细胞裂解与蛋白质提取,可以通过超声破碎设备在96孔板中有效自动化,这些设备专为高通量工作流程设计。此类仪器能够高效地提取蛋白质,并为下游高通量蛋白质纯化提供支持。目前已有多种方法成功适配微孔板形式,包括FASP、SP3和S-Trap等方法。研究表明,FASP与SP3结合使用,并配合iST,可为高通量蛋白质处理提供稳健的解决方案。
3.6.2 蛋白酶解和肽段纯化
与手动宏蛋白质组学工作流程类似,自动化蛋白处理通常包括蛋白质变性、还原、烷基化以及蛋白酶解步骤。这些步骤相对简单,可通过配备具有微量移液精度的设备和兼具加热-振荡功能的液体处理平台完成。因此,蛋白质酶解通常被认为是自动化宏蛋白质组学工作流程中复杂度最低的步骤之一。
然而,肽段纯化则面临更大的挑战。通常,这一步骤由熟练的技术人员手动操作完成,使用SPE、C18 ZipTips吸头或超滤等方法,如第3.4.4节所述。在自动化过程中,此阶段的样本异质性可能引入变化因素,使实验参数控制更为复杂。一种可行的解决方案是,将基于反相柱进行的离心步骤替换为通过移液混合反相树脂的方法。该方法已被整合至RapidAIM 2.0工作流程,并支持已建立的蛋白质组学自动化流程。例如,autoSISPROT系统提供了全吸头内样本制备功能,已证实其与自动化平台的兼容性。
3.6.3 多重标记技术
自动化样本处理与TMT标记技术的结合显著提高了宏蛋白质组学研究的通量,并加速了该领域科学问题发现的进程。然而,TMT试剂的高昂成本可能限制其广泛应用。为解决这一问题,一种策略是将TMT试剂预分装并在96孔板中干燥,这有助于减少试剂浪费并缩短实验准备时间。该方法与自动化工作流程兼容,例如RapidAIM 2.0平台中使用的工作流程,从而实现更高效的试剂利用。
尽管自动化技术在宏蛋白质组学中取得了显著进展,目前大多数系统仍处于半自动化状态,而非全自动化阶段。持续推进自动化技术的发展,对于进一步简化宏蛋白质组学工作流程、提升样本处理速度,并实现更高的分析通量至关重要。
3.7 质谱数据采集方法
宏蛋白质组的质谱分析主要依赖HP LC-MS/MS。然而质谱仪器存在一个基本限制——无法在单次运行中为样本中的所有肽段生成碎片谱图。这一局限性导致数据依赖性采集(Data-dependent acquisition, DDA)在过去25年间成为蛋白质组学的主流方法。
如3.7.1节所述,DDA策略通过选择MS1谱图中丰度最高的前体离子,并在MS2阶段对其进行碎裂,同时动态地排除已碎裂的离子,以优先选择未碎裂的目标离子。这一策略增加了肽段和蛋白质的鉴定多样性。然而,在宏蛋白质组学研究中,样本的高度复杂性对DDA在测序深度和覆盖度方面提出了巨大挑战。即使采用最新的高分辨率、高灵敏度质谱仪,DDA仍然倾向于检测高丰度离子,导致大量低丰度肽段未被表征。尽管如此,由于DDA经过广泛验证,并具备成熟的工作流程和良好的分析工具兼容性,它依然是当前最常用的方法。
如3.7.2节所述,数据非依赖性采集(Data-independent acquisition, DIA)作为新兴技术,它在预定义的质荷比(mass-to-charge, m/z)窗口内同时碎裂所有肽段离子,而非选择性靶向高丰度离子。DIA克服了DDA在肽段覆盖度和数据可重复性方面的部分局限性,因此在宏蛋白质组学中的应用日益增加。然而,DIA生成的数据更为复杂,需要先进的计算工具进行处理和分析。尽管相关工具的开发已取得进展,但在DIA成为宏蛋白质组学的常规方法之前,仍需进一步验证和优化。
DDA和DIA各具优势与局限,应根据实验目标、样本复杂性以及可用的计算资源选择合适的分析方式。
3.7.1 DDA
DDA是蛋白质组学(尤其是鸟枪法蛋白质组学)研究中最广泛使用的方法,用于鉴定生物样本中的肽段。在DDA模式下,质谱仪会动态选择指定数量丰度最高的前体离子(通常称为“topN”)进行碎裂。该优先选择机制确保每个采集周期内信号最强的离子被碎裂为更小的离子,生成MS/MS谱图,作为肽段识别的独特标记。为增强低丰度肽段的检测能力,DDA引入了动态排除机制——先前已选中的前体离子会在一定时间内被暂时排除于后续碎裂过程之外,从而提高单次运行中可分析肽段的多样性。随后,这些MS/MS谱图数据可通过蛋白质组学软件进行分析(详见第4.1.1节)。
DDA在宏蛋白质组学工作流程中具有多项优势。相较于DIA等复杂方法,其配置与分析相对简单,因此无论是初学者还是经验丰富的研究人员都能轻松使用。此外,DDA 采集的MS/MS谱图与肽段的一一对应关系降低了数据分析的计算需求,尤其是在使用高质量的蛋白质数据库时(关于蛋白质数据库的构建,请参阅第4.1.2节)。此外,DDA支持基于无标记定量(Label-free quantification, LFQ)和标记法的蛋白质相对定量,为不同实验设计提供灵活性选择(见第4.1.5节)。由于DDA在蛋白质组学领域的长期应用,大量相关的软件工具和成熟的工作流程已被开发,进一步提升了方法的可靠性与通用性。
尽管具有诸多优势,DDA仍存在明显局限。由于DDA依赖于选择丰度最强的前体离子进行碎裂,这导致低丰度蛋白可能无法被检测到(在复杂样本中尤为突出)。此外,DDA在多轮实验中常无法稳定鉴定相同肽段,导致低丰度蛋白数据缺失,给大规模定量研究带来困难。
总体而言,尽管存在局限,DDA仍是元蛋白质组学中应用最广泛、通用性最强的技术。对于需要更深入蛋白质组覆盖度或更高重复性的研究,DIA等替代方法可能提供互补优势。
3.7.2 DIA
DIA质谱技术已成为蛋白质组学中强有力的方法,可提供广泛的蛋白质覆盖度、高重现性和定量准确性。与DDA不同,DDA仅选择最强的前体离子进行碎裂,数量有限,而DIA可碎裂预设m/z窗口内的所有离子。这些窗口会在整个m/z范围内循环扫描,生成复杂的MS/MS谱图,从而提供更全面的蛋白质组视图。这一特点在宏蛋白质组学中尤为重要,因为宏蛋白质组样本包含种类繁多的肽段和低丰度蛋白,而DDA可能难以有效检测。
DIA在宏蛋白质组学研究中展现出巨大潜力。其首次应用评估见于肠道微生物组研究,并扩展至多个研究领域,包括中国白酒发酵菌种,以及基于舌苔样本的胃癌患者多队列诊断研究。近年来,随着质谱仪器的技术进步(如数据非依赖性采集-并行累积串联碎裂技术(DIA-Parallel accumulation serial fragmentation, DIA-PASEF)及Orbitrap Astral)DIA的灵敏度和分辨率显著提升,使其在高度复杂的微生物群落中实现更深度的蛋白质组覆盖。
相较于DDA,DIA的核心优势在于能捕获更广泛的肽段,从而实现更深的蛋白质组覆盖并提升低丰度蛋白检出率。另一突出优势是其跨样本的高重现性——由于受电离效率波动影响较小,这一特性使其特别适用于大规模定量研究。
尽管DIA具有诸多优势,但其数据分析仍面临挑战。DIA生成的复杂MS/MS谱图需借助先进计算工具和专业分析技能进行解析(详见第4.1.1节)。此外,由于DIA会同时碎裂特定m/z窗口内的所有离子,因此所得MS/MS谱图相比DDA具有更高的复杂性,且对单个肽段的特异性较低。这种特异性的降低可能影响DIA在解析肽段精细结构或序列信息方面的能力,限制了DIA在PTM研究或高相似度肽段区分等需精准表征领域的应用。这些权衡凸显了针对特定研究目标优化DIA工作流程的必要性。
尽管如此,DIA的快速发展使其成为宏蛋白质组学研究的潜力工具,为全面探索微生物群落功能景观提供了所需的深度与重现性。
3.7.3 优化HPLC和MS分析的关键参数
优化HPLC和MS方法对于在宏蛋白质组学工作流程中获得高质量数据至关重要。以下每个参数在确保准确的肽段分离、鉴定和定量方面都起着重要作用。由于宏蛋白质组学相较于标准蛋白质组学具有更高的复杂性,因此需要针对许多参数进行特定调整。
i)分析柱质量、梯度和流速
肽段通常通过HPLC进行分离,该系统直接与MS联用,使用商业或自制的分析柱进行分离。分离过程通过乙腈(Acetonitrile, ACN)浓度递增的流动相完成。对于使用自制色谱柱的实验室,严格的质量控制检查至关重要,以确保色谱柱性能的稳定性,详见第3.7.4节。
宏蛋白质组学由于其样本固有的复杂性,相较于单物种蛋白质组学带来了更大的挑战。为应对这一问题,通常采用5%–35%(以80% ACN 计)或5%–30%(以100% ACN计)的梯度,在1–2小时内完成肽段的洗脱。然而,在特定实验条件下,可能需要进行相应调整。例如,化学标记的酶解产物由于其疏水性增强,通常需要更高终浓度ACN的洗脱梯度,以确保肽段被完全洗脱。
高效梯度设计对优化运行时间、实现均匀的肽段洗脱分布至关重要。由于梯度起始与结束阶段肽段洗脱量较少,对梯度进行合理调整可以提高分离和检测效果。准确的肽段定量需要确保每个LC色谱峰具备足够的采样点,因此,短梯度(例如10分钟梯度)通常不适用于DDA模式下的宏蛋白质组学。有关梯度优化的详细教程可参考蛋白质组学及宏蛋白质组学领域的相关文献。
液相流速通常设定在200–300 nL/min之间。近年来,利用提高液相色谱流速以加快样品分析周期的策略,逐渐受到研究者的广泛关注。然而,提高流速会降低检测灵敏度。为了应对这一问题,可以增加样品分析量,或使用二甲基亚砜(Dimethyl sulfoxide, DMSO)提升信号强度,从而使高液相流速更适用于宏蛋白质组学工作流程。
ii) DDA工作流程中的质谱参数设置
优化MS参数对获取高质量宏蛋白质组学数据至关重要。尽管初学者通常无需自行配置MS参数,但了解关键参数优化步骤可以为数据解析和故障排除提供有效的背景信息。
为了确保精确的质量测量,必须定期校准质谱仪,以提高肽段鉴定和定量的可靠性。此外,优化离子源参数(如源温度、流速和雾化气压)以增强电离效率并最大化信号强度。具体的优化步骤会根据质谱仪类型有所不同,如飞行时间 (Time-of-flight, TOF) 或 Orbitrap 质谱。这些质谱仪器的关键参数包括扫描范围、分辨率和扫描速度,需精细调整以确保精确的质量测量,并能有效分辨相邻的肽段分子。同时,肽段碎裂的碰撞能量也需要仔细设定,以生成高质量的碎片谱,进而用于肽段的准确鉴定。
动态排除是DDA工作流程中的一个重要参数,它需要根据色谱梯度与峰宽仔细校准。该设置通过暂时排除已碎裂肽段防止重复碎裂,从而增加肽段多样性。但在宏蛋白质组学研究中,此方法面临特殊挑战。许多研究人员依赖谱图计数进行相对定量,特别是在宏蛋白质组学数据集中,由于细胞数量和不同群体之间的总蛋白质含量存在显著差异,谱图计数具有较好的稳健性。然而,动态排除可能导致高丰度肽段的谱图采集次数减少,从而导致谱图计数低于预期,进而影响定量的准确性。随着现代高分辨率质谱仪的发展,扫描速率的提高和分辨能力的增强使得肽段丰度与肽段-谱匹配数(Peptide-spectrum matches, PSMs)之间的相关性降低。因此,动态排除时间的设定需要在确保高质量碎片谱图的同时,又要最大化分析肽段的多样性。在宏蛋白质组学研究中,谱图计数与基于MS1信号强度的定量方法(如曲线下面积(Area under the curve,AUC))之间的选择仍然存在一定的争议。
在DDA工作流程中,前体离子的隔离窗口宽度是一个关键的优化步骤。较宽的隔离窗口(可达2 Da)能捕获更多离子,进而获得更高质量的MS谱图。但这也增加了生成嵌合谱图的风险,即多个前体离子的碎片可能混合在一起,从而增加肽段鉴定的复杂性。相反,较窄的隔离窗口(如 0.7 Da)能够减少嵌合谱图的产生,但会限制隔离离子数量,这可能会影响信号强度。在宏蛋白质组学研究中,由于特定质量范围内前体离子的密度高和多样性大,即使使用较窄的隔离窗口也可能捕获多个离子。随着质谱技术的进步,尤其是扫描速度的提升,使得DDA模式支持设置更高的topN值,从而在一次分析中获取更多的碎片谱图,有助于应对这一挑战。
iii) DIA工作流程中的质谱参数设置
优化DIA工作流程需要对多个关键参数进行精准调整,以确保准确且全面的肽段鉴定。质量隔离窗口的宽度尤为关键,较窄的窗口(如 2 m/z)能够提供更高的分辨率和更精确的碎片谱图,这对于解析复杂的肽段混合物至关重要。然而,较窄的窗口会降低蛋白质组的覆盖度,因为每个循环中被隔离的前体离子数量较少。因此,平衡分辨率与覆盖度是DIA优化的核心挑战。最近的技术进展,如Orbitrap Astral质谱仪,支持超窄的隔离窗口,同时保持较高的扫描速度,有效地弥合了DDA和DIA方法之间的差距。
除了调整隔离窗口,优化碰撞能量也是确保生成高质量碎片离子的关键。同时,仔细校准色谱条件(包括梯度时长与流速)使其与DIA周期时间匹配,以确保在肽段洗脱峰的整个过程中获得足够的数据采集点,从而实现精确的肽段鉴定和定量。随着DIA工作流程的迅速发展,宏蛋白质组学中的DIA技术能够在复杂的微生物组样本中实现更高的分辨率和更全面的蛋白质组覆盖度。关于这些优化策略的详细指南,可以参考近期关于DIA方法学的进展研究。
3.7.4 LC-MS/MS的质量控制
质控流程第一步是注入空白无样本溶剂,旨在监测系统背景污染并排除外源性干扰。理想情况下,空白样品上机应产生极少量的鉴定结果,这可以通过目视检查或数据库检索进行验证。污染源可能包括HPLC系统中使用的输送溶剂,因此需严格监控这些溶剂。随后需利用质谱仪分析已知肽段混合物标准品,例如细胞色素C或牛血清白蛋白(Bovine serum albumin, BSA)酶解产物,以确认仪器的正常校准和性能。此类简单的肽段混合物样本可用于测试HPLC性能,而更复杂的肽段混合物样本(如HeLa细胞酶解产物)可评估质谱仪分析复杂样本的能力。此外,还可以注入代表性的微生物组肽段样品,以优化液相梯度参数,此类标准样品应在质谱仪的整个运行过程中定期进样。此外,使用参考微生物组样本作为阳性对照有助于验证蛋白提取方案的效率,确保提取方法能够可靠地捕获样本中代表性蛋白质,这对于宏蛋白质组学研究尤为关键。对复杂标准品质谱数据进行数据库检索可用于监测PSMs数量、肽段及蛋白质的鉴定数量。长期跟踪这些指标,有助于检测质谱仪的性能,并提示质谱仪器是否需要进行清洁或重新校准。
在LC-MS/MS运行期间中,必须密切监控已知峰的保留时间,显著的偏移可能提示存在色谱柱堵塞、连接器泄漏或阀门磨损等问题。柱后压也可作为发现潜在问题的指标进行监测。同时,还需评估峰形的对称性和尖锐度,峰尾或峰宽异常可能提示色谱或电离效率存在问题。信号强度是另一个重要的监测参数,若信号强度显著低于预期值,可能表明质谱仪灵敏度降低或电离异常。
质谱仪器运行结束后,需仔细检查每个原始质谱数据,以识别潜在问题。如果某次运行失败,应立即将该样本重新进行分析,以避免因延迟时间导致批次效应。总离子流(Total ion current, TIC)色谱图提供了有关仪器性能的关键信息,应检查是否存在异常峰或基线噪声,可能提示污染或硬件问题。基峰色谱图可辅助评估液相分离效果。比较TIC与基峰强度比值也具有参考价值,较高比值通常反映样本复杂度增加或色谱性能下降。跨样本的保留时间以及峰强度应保持一致,提示数据的良好重复性。其他质控方法,如主成分分析(Principal component analysis, PCA)或热图,可帮助识别不同样本运行之间的差异,确保数据质量。
蛋白质鉴定和定量后收集的各项指标对质控评估同样关键。例如,PSMs鉴定数量与MS2谱图总数的比例,即PSMs鉴定率,是评估质谱数据质量的关键指标。以Q-Exactive质谱仪为例,在使用1小时分析梯度、优化参数和高质量样品准备下,宏蛋白质组样本的鉴定率大约为50%,即在1% FDR过滤后,50%的谱图可匹配鉴定到肽段序列。需要注意的是,对于土壤等复杂环境的样本,PSMs鉴定率通常较低。在分析高质量QC样本时,必须使用相同的LC-MS/MS方法,因鉴定率高度依赖于仪器性能和样品质量。
在持续数周的大规模项目中,保留时间偏倚和信号衰减是常见现象。为了减少此类性能波动带来的系统性偏差,建议在分析过程中进行样本分块和随机化分析策略。在LC-MS/MS各环节实施严格的质量控制对维持数据可靠性与一致性至关重要,标准化的质控样品可作为质谱仪长期性能的有效基准。
现可用多种质控工具评估LC-MS/MS数据质量,例如MaCProQC、QCloud2和Rawtools。这些工具提供了从性能指标追踪到数据质量聚类分析等功能。近年来,人类蛋白质组组织标准化倡议(Human Proteome Organization Proteomics Standards Initiative, HUPO-PSI)质控工作组推出了mzQC文件格式,这是一个基于JSON的标准格式,旨在规范质控指标报告与交换。为了促进应用,他们还开发了Python(pymzqc)、R(rmzqc)和Java(jmzqc)等开源软件库。这些库使研究者能将mzQC整合至蛋白质组学、代谢组学等质谱应用流程,确保数据质量评估一致性,并促进不同分析平台之间的互操作性。
3.7.5 数据管理与数据共享
高效的数据管理与共享对推动宏蛋白质组学研究至关重要,它能够确保数据的完整性、可重复性与及促进协作。一个完善的数据管理计划应包括安全冗余的存储策略,以防止数据丢失,尤其是在进行大规模、长期研究时。此外,实施原始数据和处理数据的版本控制,有助于系统地追踪更新和重新分析数据,进而提高研究的可重复性和透明度。
遵循HUPO-PSI制定的标准,对于确保数据的一致性和互操作性极为关键。HUPO-PSI制定了蛋白质组学数据表示的标准,旨在促进数据的比较、交换和验证。使用标准化格式,如质谱数据采用mzML、鉴定结果采用mzIdentML、引用公共蛋白质组数据集质谱时采用通用谱图标识符USI,能够确保跨平台与工具的兼容性,从而简化协作并提升数据利用效率。
元数据在确保数据集的可解释性、可重复性和可比较性方面发挥着重要作用。完整全面的元数据应涵盖样本来源、制备流程、仪器参数和数据处理工作流程等信息,最好采用标准化本体,如PSI-MS本体。在蛋白质组学中,这些信息通常以蛋白质组学样本和数据关系格式(Sample and Data Relationship Format for Proteomics, SDRF-Proteomics)进行收集,这是一种结构化的、制表符分隔的格式,用于描述样本与数据文件之间的关系,并能反映蛋白质组学实验流程。lesSDRF等工具提供了便捷的用户界面,帮助在SDRF格式中标注元数据,促进数据的标准化进程。鉴于微生物环境的特殊复杂性,宏蛋白质组学倡议正在开发适用于宏蛋白质组学的SDRF-Proteomics模板,因为现有针对单一物种蛋白质组学的格式无法完整描述微生物数据的细微差异。标准化的元数据不仅支持计算分析,还为机器学习模型提供了结构化的输入,从而推动领域内的可重复性和标准化。
将数据和元数据提交存储在国际公认的蛋白质组学交换库(ProteomeXchange)如PRIDE中,符合可查找、可访问、可互操作和可重用(Findable, Accessible, Interoperable, and Reusable, FAIR)原则,能够促进开放科学和创新。此类公共数据库向研究界开放数据访问,支持研究成果的验证、系统评估以及大规模数据分析。宏蛋白质组学中的数据共享有助于基准研究、新型解析工具的开发以及广泛结论推导,从而显著提升该领域的协作潜力和影响力。
4. 宏蛋白质组学数据的计算分析
4.1 肽段鉴别、蛋白质推断和定量
在获得质谱数据后,下一步是鉴定样品中的肽段。这一过程通过解析质谱数据中的碎片特征来确定肽段的氨基酸序列。该步骤通常借助检索引擎(通常整合于蛋白质组学软件包中,详见第4.1.1节)实现。检索引擎将实验获得的质谱数据与理论蛋白质序列数据库进行比对,成功的肽段-谱图匹配高度依赖于合适数据库的选择或构建(如第4.1.2节所述)。随后,检索引擎应用假发现率(False discovery rate, FDR)阈值来过滤潜在的假阳性结果(见第4.1.3节)。这些经过过滤的肽段随后用于蛋白质推断(见第4.1.4节)和定量(见第4.1.5节)。上述章节均聚焦DDA质谱数据的处理,第4.1.6节则专门讨论用于分析DIA质谱数据分析工具。
4.1.1 基于蛋白质组学检索引擎的肽段鉴定
鸟枪法宏蛋白质组学实验会生成大量的MS1和MS2谱图数据,这些数据是下游分析的基础。随着高通量质谱技术的进步,数据集的规模可达数千甚至数百万张谱图,使得人工解读变得不切实际。检索引擎成为数据解析与肽段鉴定的核心工具。肽段鉴定主要依赖于三种策略:(i)序列数据库检索,通过将实验谱图与来自蛋白质或肽段序列数据库的理论谱图进行匹配;(ii)从头测序,直接从原始数据中的谱图推断肽段序列,无需参考数据库;(iii)谱图库检索,通过将实验谱图与已验证的谱图库进行对比。为了提高肽段鉴定的准确性和可信度,这些方法常辅以后处理步骤,如第4.1.3节所述。此外,大多数蛋白质组学软件包还将肽段鉴定、蛋白质推断、定量整合,相关内容将在第4.1.4节和第4.1.5节中讨论。部分宏蛋白质组学软件还整合了分类与功能分析模块,详见第4.2节。
i)蛋白质序列数据库检索
蛋白质数据库检索算法是解读质谱数据的基础,尤其在宏蛋白质组学中,微生物群落的复杂性带来了显著的分析挑战。这些算法将谱图数据与从蛋白质序列数据库生成的理论谱图进行比对匹配。数据库检索引擎首先选定蛋白质序列数据库,这些蛋白序列经过计算机模拟酶切。根据这些酶切序列生成理论谱图,并与质谱实验中获得的谱图进行对比。每个PSM会被赋予一个相似度评分,检索引擎根据评分与肽段特性对潜在PSM排序筛选。具体评分算法因检索引擎而异,这些差异可能影响灵敏度和特异性。各检索引擎评分机制详见该综述。
每个数据库检索引擎都有其优点和局限性,包括处理速度、输入输出格式兼容性、对后处理工具的支持以及用户友好性等方面的差异。这些因素会显著影响宏蛋白质组学工作流程的表现,因为复杂大规模数据集需借助高效可靠的分析工具。相关工具应用评述可参考这篇综述。以下是一些在宏蛋白质组学研究中常用的数据库检索引擎与软件的介绍:
SearchGUI支持同时调用X!Tandem、Comet、Andromeda、OMSSA、Sage等互补算法。其配套工具PeptideShaker可无缝导入SearchGUI输出,并提供了一个全面的用户友好界面,用于结果解析和可视化并支持直接导出至Unipept平台实现下游分类与功能分析。MaxQuant内置的Andromeda,因其易用性和MS1谱图定量能力广受青睐,用户社区活跃(含年度用户会议与专属论坛)。Mascot(Matrix Science)和Proteome Discoverer(Thermo Fisher Scientific)是用户基数庞大的商业化工具。采用MSFragger的FragPipe和pFind整合开放搜索策略,通过识别PTMs来提升灵敏度。Sipros 、ProteoStorm 和COMPIL 2.0 专门为宏蛋白质组学设计,但用户友好性不及主流软件。Sage 和MSFragger 等工具利用先进的谱图和序列索引策略,显著加速数据库检索,因此在提高宏蛋白质组学分析速度方面具有巨大潜力。
研究者若需集成化解决方案,可选下列软件套件简化宏蛋白质组学工作流:Galaxy for Proteomics(Galaxy-P)是一个多功能平台,提供了许多针对宏蛋白质组学的工具,包括数据库构建、发现分析、验证、定量和统计分析,其公共网关与Galaxy培训网络资源助力用户便捷开展分析。MetaProteomeAnalyzer(MPA)软件套件提供了蛋白质序列数据库构建、数据库检索、蛋白质分组、注释和结果可视化等模块,适合各层次研究者。MetaLab 是一个集成的数据处理工作站,包括样本特异性数据库生成、肽段鉴定、分类学和功能分析以及丰度定量工具。其开放搜索策略能够支持全面PTMs分析,并提高灵敏度。此外,MetaLab提供了基于宏基因组组装基因组(Metagenome-Assembled Genome, MAG)数据库的分类学分析流程,可以通过肽段与基因组关联,相较于传统的最小公共祖先(Lowest common ancestor, LCA)方法具有更高的特异性。
在这些工具中,选择合适的检索参数对于获得可靠且有意义的结果至关重要。关于修饰、酶特异性和质量偏差等参数的设定将显著影响PSMs的鉴定。以下是几个关键的考虑因素:
肽段修饰选择:区分实验步骤引入的修饰与生物学修饰非常重要。常见的固定修饰,如半胱氨酸的烷基化修饰,通常应用于所有肽段(第3.4.4节)。可变修饰,如甲硫氨酸氧化修饰,用于探索生物学相关的修饰。然而,包含过多的可变修饰可能会过度扩展检索空间,从而减少鉴定率,建议仅纳入关键生物修饰。
特异性酶切位点和漏切数目:正确设置酶切位点与允许漏切数影响可检测肽段范围。以常用胰蛋白酶为例,通常允许一个或两个漏切数能平衡酶切不完全与搜索空间过度扩展。在某些情况下,半特异性或非特异性酶切参数设置可能会有用,但可能会延长处理时间并降低鉴定率。
质量偏差:质量偏差设置应与质谱仪的分辨率能力相匹配。例如,在高分辨率的Q Exactive质谱仪上,采用高能碰撞解离(Higher-energy collisional dissociation, HCD)碎裂时,设置10 ppm的前体离子质量偏差和0.02 Da的碎片质量偏差可以有效平衡精度与计算算力。
合理的参数设置能平衡灵敏度与特异性,生成准确反映样本生物学特征的高质量数据。参数调整应考虑质谱仪类型、样本复杂性和具体研究目标。
ii)de·novo测序
从头(De·novo)肽段测序无需依赖蛋白质序列数据库进行谱图匹配,而是直接为MS/MS谱图分配氨基酸序列。这种方法提供了一种无偏倚的肽段检测方法,不受蛋白质序列数据库质量和完整性的限制。近年来已开发多款从头测序算法,包括 PEAKS、Casanovo、PepNovo,以及新近推出的π-HelixNovo 、metaSpectraST 和NovoBridge。
从头测序能够在无先验知识条件下,灵敏且准确地推测微生物群的分类组成和功能特征。它还具有识别微生物群中未测序物种的潜力。此外,从头测序可用于评估蛋白质序列数据库在宏蛋白质组学研究中的完整性与适用性。最近的一项综述系统探讨了从头测序在宏蛋白质组学中的最新进展和发展机遇,强调了其在未测序物种检测及微生物群落功能解析方面的巨大潜力。
尽管从头测序具有广阔的应用前景,但仍需要对相关工具进行系统化基准测试,以评估其在宏蛋白质组学研究中的适用性。尤其是,大多数从头测序分析工具和方法仍需借助数据库信息,例如用于辅助筛选肽段或从已鉴定的肽段中获取信息。因此,评估这些工具在灵敏度、准确性和通量方面的表现至关重要,以确保它们能有效处理微生物组研究中复杂多样的数据集。
iii)谱图库检索
谱图库检索引擎的运行原理与数据库检索类似,但不同之处在于它直接将实验MS/MS谱图与预先构建的、经过验证的谱图库进行比对。这些谱图库可由先前通过复杂肽段混合物分析和传统序列数据库检索获得的MS/MS谱图构成,或是利用深度学习算法预测生成。相较于序列数据库搜索,谱图库搜索可整合更多参数,例如液相保留时间与谱图内碎片峰的相对强度,从而提升肽段鉴定的准确性和置信度。
近年来,基于AI的工具(如MS²PIP和Prosit )的开发,使得研究人员能够从蛋白质序列数据库生成高质量的谱图库成为可能,进一步拓展了谱图库检索的应用范围,使其能够针对特定实验生成高质量的预测谱图库。此外,近年来针对DDA数据设计的谱图库检索工具(如Mistle和Scribe)也已应用于宏蛋白质组学研究。
谱图库检索提供了一种快速高效的策略,可将肽段序列与MS/MS谱图匹配,在检索速度和精度方面通常优于传统的数据库搜索,尤其是在使用高质量谱图库时。然而,尽管谱图库检索展现出巨大的潜力,其在宏蛋白质组学研究中的适用性仍需进一步评估,尤其是在面对高度复杂的微生物数据集时,仍需优化其可用性和有效性。
4.1.2 数据库构建或选择
对于单一生物的蛋白质组学研究,蛋白质序列数据库的构建相对简单,可直接根据从该生物体的基因组导出。然而,在宏蛋白质组学研究中,由于微生物群落的复杂性、生物种类的多样性及未知蛋白的存在,数据库的选择或构建面临严峻挑战。选择或生成合适的数据库至关重要,因为数据库必须在全面性和样本特异性之间取得平衡。不完整的数据库可能导致蛋白漏检或错误识别,而数据库规模过大则可能降低分析灵敏度,并抬高FDR,详见第4.1.3节。
一个理想的宏蛋白质组数据库应兼具全面性和特异性。全面性意味着数据库应尽可能包含样本中所有潜在的蛋白质,以避免因蛋白序列缺失导致假阴性结果,降低肽段和蛋白质的鉴定率;特异性则要求数据库剔除不应出现在样本中的序列,否则可能因无关序列导致随机匹配增多,从而提高FDR,影响肽段(和蛋白质)的准确鉴定(详见第4.1.3节)。此外,宏蛋白质组学分析通常需考虑样本处理过程中引入的污染物,如残留胰蛋白酶、BSA污染或实验操作引入的角蛋白。为避免这些污染物干扰蛋白鉴定,可整合纳入污染物信息的“常见外源蛋白数据库”(common Repository of Adventitious Proteins, cRAP,https://www.thegpm.org/crap/),以便准确识别这些污染物并防止与样本中其他蛋白质的错误鉴定。
数据库的构建依赖于对微生物群落组成的先验知识。这类信息可来源于文献调研、16S rRNA扩增子测序或宏基因组/宏转录组技术等不同途径。其中,文献综述提供的信息相对有限,而宏组学技术则能更全面地表征群落组成。此外,根据样本来源环境,可能需在数据库中纳入宿主或膳食蛋白质。尽管加入这些蛋白可提高鉴定率,但同时也会增加数据库体量及复杂性,可能影响分析效率。由于大型数据库往往包含高度相似的序列,这可能进一步加剧蛋白推断问题(详见 4.1.5 节)。此时可采用序列聚类算法或蛋白质分组工具来整合冗余蛋白序列,同时保留必要的分类学和功能注释。
数据库类型的选择取决于样本类型、微生物群落的已知程度及可用资源。不同类型的数据库各具优势和局限性(详见表2),常见类别包括公共数据库、参考目录和宏组学数据库,具体如下。
i)公共数据库
公共数据库(如UniProtKB和NCBI RefSeq)提供了广泛的蛋白质序列参考集合。然而,这些未经筛选的数据库通常缺乏特异性,包含大量无关序列,导致鉴定率下降并增加FDR(详见第4.1.3节)。此外,公共数据库往往对研究充分的微生物存在偏向,例如模式生物或病原体,以及对特定环境或系统(如临床和人类样本)进行了深入研究。因此,对于研究较少的环境微生物群落,这些数据库通常是不完整的。基于16S rRNA分析结果对这些数据库进行筛选可以提高特异性,但16S rRNA测序的分辨率有限,往往需要纳入整个属或多个物种,而无法实现菌株水平的特异性。
ii)参考目录
参考目录是针对特定环境或系统精心整理的蛋白质序列集合,如人类肠道、牛瘤胃和小鼠肠道等参考目录。这些目录通常整合了分离培养的微生物数据和宏基因组研究数据。相比公共数据库,参考目录体量更小、更具针对性,但对于宏蛋白质组分析而言,仍可能过于庞大。此外,参考目录通常整合多个样本的数据,包括不同个体和研究的结果,与研究样本本身无直接对应,因此也被称为非匹配宏组学数据库。即便是相似环境的样本,在物种组成和菌株多样性上仍可能存在较大差异,导致参考目录可能存在不准确、不完整和某些子样本过度表征等问题。与公共数据库类似,参考目录的特异性可通过结合微生物群落的先验知识(如16S rRNA分析结果)优化,仅保留与样本最相关的序列。
此外,为减少数据库的规模并提高数据利用率,研究人员提出了一些数据库优化策略,例如两步法搜库策略、MetaPro-IQ和MetaLab等迭代流程及其他方法。这些方法已广泛应用于宏蛋白质组研究,并能提高PSMs和肽段鉴定数量。然而,研究表明,其中部分方法虽能提高鉴定数量,但也可能显著增加假阳性鉴定情况,导致FDR估计值偏高。因此,在得出生物学结论前,需谨慎采用这些方法,并辅以额外验证。
iii)(匹配的)宏组学数据库
宏组学数据库是基于与宏蛋白质组学分析同一样本的宏基因组和/或宏转录组数据进行构建,因此具有最高的样本特异性。这类数据库能够准确反映样本的物种组成及菌株多样性。然而,构建高质量的宏组学数据库需要较高的测序深度、成本、计算资源以及专业技术。尽管具体构建细节超出本文范畴,但已有文献对此进行了详尽阐述。简而言之,构建宏组学数据库通常包括四个关键步骤:测序、组装、分箱和注释。
为了构建适用于宏蛋白质组学分析的全面数据库,测序深度必须足够高,以便充分捕获群落的复杂性。宏组学数据库的核心优势在于能够精准解析样本内的物种及菌株多样性,并直接关联基因组信息与鉴定出的蛋白质。这一过程需要通过分箱进行基因组重建,即基于共有特征将重叠群(contigs)聚类为宏基因组组装基因组(Metagenome-Assembled Genomes, MAGs)。然而,由于微生物群落的复杂性及测序深度的限制,部分MAGs可能仍不完整。因此,高质量的宏组学数据库需同时包含分箱和未分箱的序列,以最大限度保留信息。
基因组重构完成后,需对MAGs和重叠群进行物种注释,并预测蛋白质序列或开放阅读框(Open-reading frames, ORFs),随后进行功能注释。这些步骤所采用的工具和资源取决于研究目标。尽管宏组学数据库具有较高的特异性,但仍可能因测序深度不足或无法从样本中捕获所有相关MAGs而导致数据库存在一定不完整性。该问题可通过探索性16S rRNA基因测序评估宏基因组分析所需的最优测序深度,从而在一定程度上加以缓解。
将宏基因组数据与宏转录组数据结合,可进一步提升数据库的质量和特异性。由于宏转录组学侧重于mRNA的解析,其捕获的是微生物群落的活跃功能部分,从而提供基于基因表达的视角,使其与宏蛋白质组学关注的功能信息更紧密匹配。
表2. 宏蛋白质组学数据库类型的比较:公共数据库、参考目录与多组学数据库
颜色标识偏好度:绿色-优选,黄色-中等,红色-不推荐。
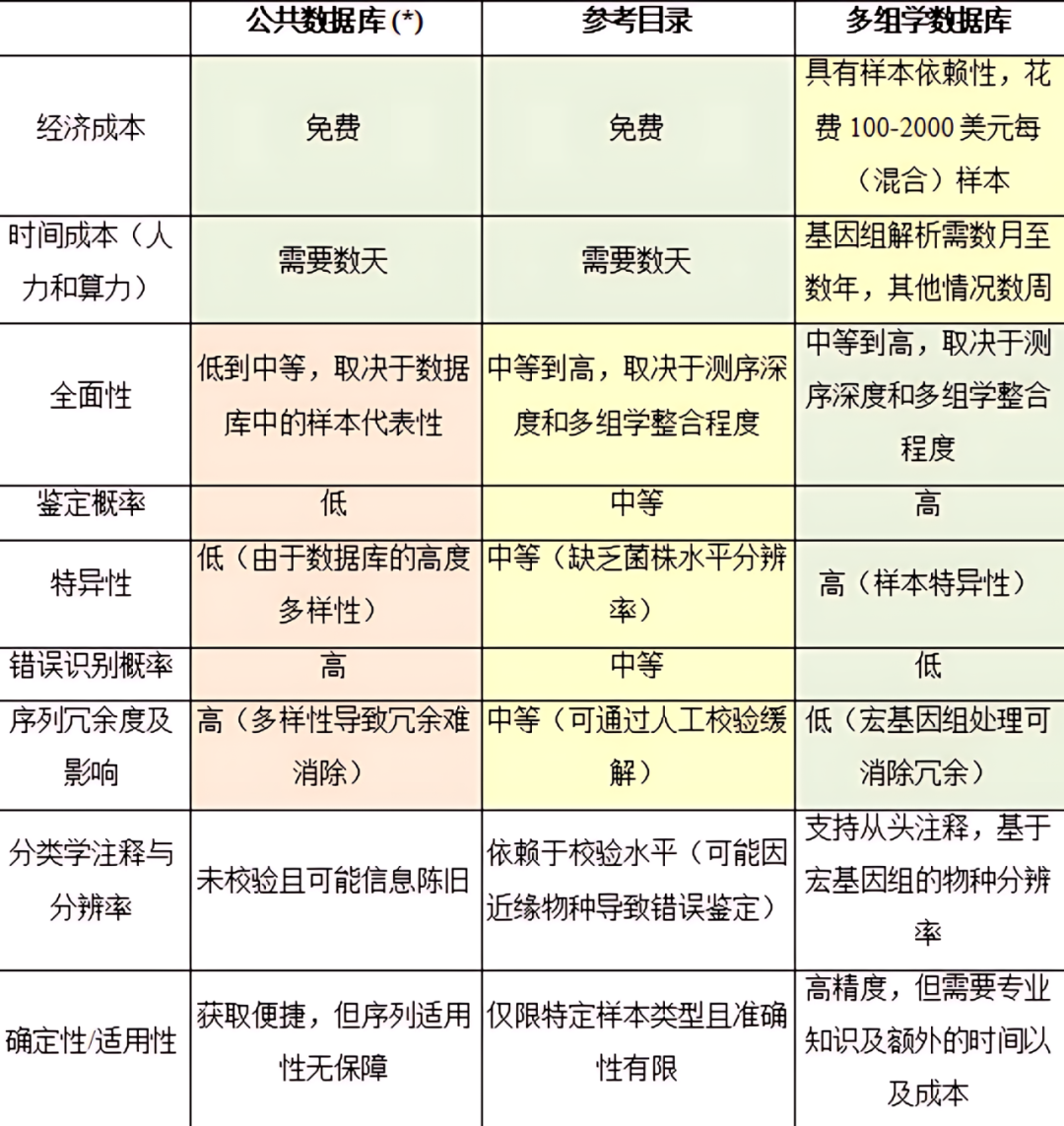
(*) 限制性公共数据库在特异性和序列冗余度指标方面与参考基因组目录特性相似。
4.1.3 PSM FDR质控
在肽段鉴定过程中,获取可靠的PSM是关键步骤之一。在获得PSM后,它们会根据搜索引擎的评分函数进行评估,并保留每个谱图中匹配得分最高的PSM,即理论谱图与实验MS/MS谱图最匹配的肽段序列。然而,无论采用何种评分算法,都难以完全避免错误匹配,因此需要控制假阳性匹配情况。
在(宏)蛋白质组学研究中,控制假阳性最常用的方法是目标-诱饵策略。该方法首先对目标数据库中的蛋白质序列进行计算模拟酶切,以生成理论肽段。随后,对诱饵数据库中的反向或重排序列进行相同处理,确保诱饵肽段不具备生物学真实性且不存在于样本中。在检索过程中,实验谱图会与目标-诱饵数据库中的目标和诱饵序列进行匹配。最终,产生靶向或诱饵PSM标记。最终结果中诱饵PSM占比可作为FDR估计值,计算公式为:诱饵PSM数量 / 得到的PSM总数(图4)。在蛋白质组学和宏蛋白质组学实验中,FDR通常控制在1%,但对于土壤微生物组等高度复杂的样本,为了确保获得足够的鉴定数量以进行生物学分析,FDR阈值可放宽至5%。
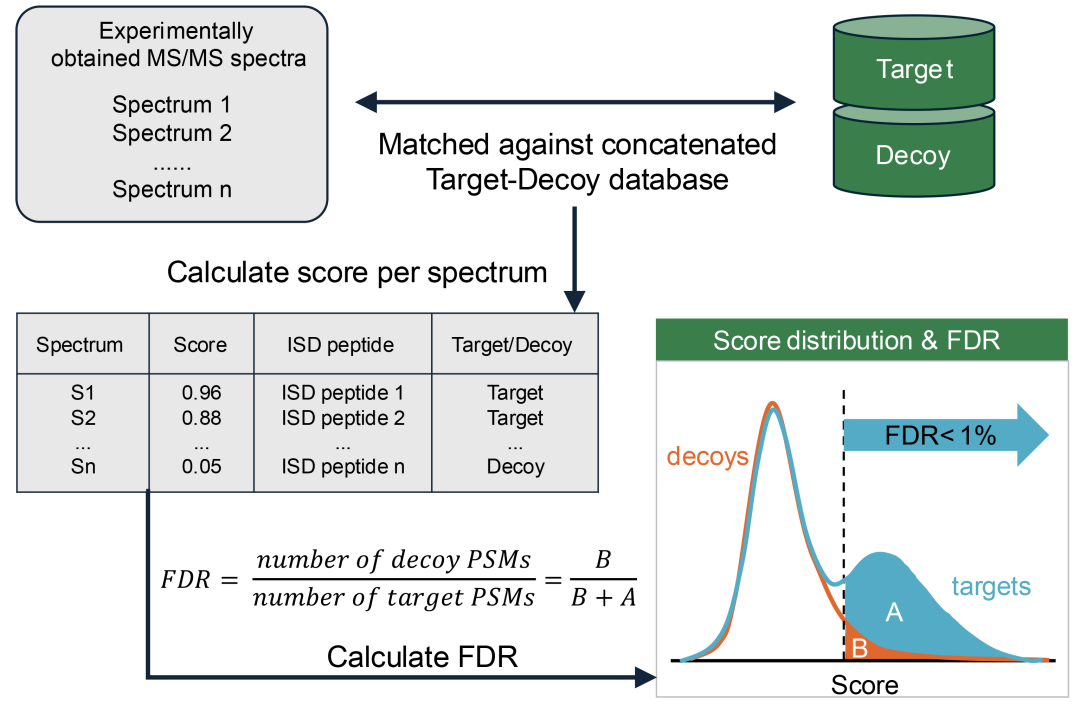
图4. 目标-诱饵分析原理及FDR计算
(上图)实验获得的MS/MS谱图与计算生成的目标/诱饵蛋白序列数据库谱图进行匹配。(中图)每个谱图保留得分最高的匹配,并记录对应的理论酶切肽段序列及目标/诱饵标签。(下图)基于得分分布筛选真实PSMs。通过FDR控制假阳性,其计算公式为诱饵PSMs数量除以目标PSMs数量(图中表示为B区面积除以A+B区面积之和)。本图改编自[253]。
宏蛋白质组学的特殊使FDR控制更加复杂。宏蛋白质组学通常需要大规模、高多样性蛋白质序列数据库。但这会扩大检索空间,从而导致目标和诱饵 PSM 评分分布之间的重叠度增加。这种重叠会降低FDR估算的分辨率,因此需要谨慎构建数据库,以减少无关序列的干扰(见第4.1.2节讨论)。过度庞大的非特异性数据库会增加目标和诱饵序列的随机匹配,从而提高FDR,减少高置信度肽段鉴定。反之,数据库若过于严格,可能会导致真实的目标序列被排除,从而增加假阴性情况,并降低蛋白质组的覆盖度。因此,在数据库的特异性和全面性之间实现最佳平衡至关重要,以最大程度减少由诱饵匹配导致的假阳性,同时尽可能多地识别目标肽段,从而确保有效的FDR质控。
宏蛋白质组学工作流程通常依赖于先进的后处理工具,以提高肽段鉴定的准确性和置信度。例如,MS²Rescore通过整合Percolator的搜索引擎依赖性特征、MS²PIP和DeepLC的衍生特征,优化PSM评分。这些预测特征与Percolator的半监督机器学习模型结合后,可增强靶向与诱饵PSM的区分度,实现更精准的FDR估计。此类优化不仅能提升肽段鉴定率,还可提高下游分类与功能分析的可靠性,对复杂微生物组数据集尤为关键。
在宏蛋白质组学研究中,由于样本通常包含数千种微生物,微生物群落固有的复杂性与多样性进一步放大了FDR控制难度。通过谨慎的数据库构建(第4.1.2节)、检索阶段的严格FDR控制以及先进的后处理技术相结合,才能确保肽段与蛋白质鉴定的可靠性,从而使宏蛋白质组数据能够提供有意义的生物学见解。
4.1.4 蛋白质推断
蛋白质推断是自下而上蛋白质组学中的核心挑战,其目标是在基于串联质谱鉴定出的肽段基础上,推测样本中实际存在的蛋白质。这一过程的复杂性在于,鉴定出的肽段可映射至蛋白质数据库中多个蛋白质或蛋白质异构体。在复杂样本(如微生物群落样本)中,多个物种可能含有高度相似的同源蛋白,导致难以准确推断样本中真实存在的蛋白。
为了解决这一问题,通常采用蛋白分组策略,以生成更便于下游分析的蛋白(亚)组列表。然而,不同的蛋白分组方法有所不同,如图5所示,这些方法通常由检索引擎执行。因此,研究者需需验证搜索引擎的默认设置以理解其采用的分组策略,必要时调整以匹配研究假设。蛋白推断的两种主要方法分别是奥卡姆剃刀原则和反奥卡姆剃刀原则。
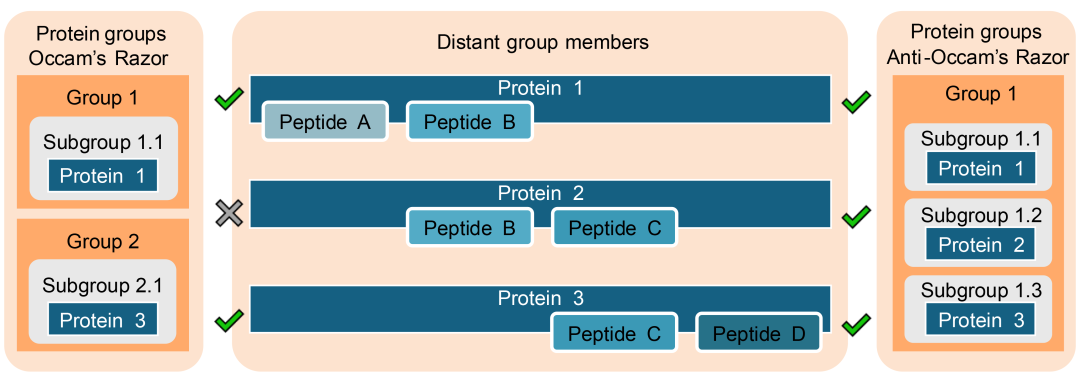
图5. 蛋白质(亚)组分组策略的实践案例
本示例图中存在远缘成员(蛋白质1与蛋白质3无共享肽段),应用简约原则分为两个蛋白质小组。而在应用反奥卡姆剃刀原则时,蛋白质2仍然作为一个单独的亚组进行保留。
奥卡姆剃刀原则基于最大简约性,即选择能够解释所有观察到肽段的最小蛋白集合。然而,该方法会舍弃无唯一肽段匹配的蛋白,可能导致部分蛋白的分类学信息和功能信息丢失。奥卡姆剃刀原则特别适用于单一物种样本或靶向蛋白质组学实验,在这些研究中,核心目标是降低复杂性。
相比之下,反奥卡姆剃刀原则采用包容性策略,保留所有能匹配至少一个肽段的蛋白质,而不考虑这些肽段是否与其他蛋白质共享。该策略对复杂宏蛋白质组样本尤为有利,因为其目标是尽可能捕获蛋白多样性。因其旨在最大限度捕获蛋白质多样性。通过包容性策略,可避免遗漏仅含少量唯一肽段的不同物种蛋白质,从而更全面地呈现微生物群落特征。然而,这种方法的缺点是会增加所得蛋白列表的复杂性。
选择奥卡姆或反奥卡姆原则后,蛋白质可进一步分为蛋白质组或蛋白质亚组。蛋白质组即共享至少一个肽段的蛋白质集合,提供更宽泛的潜在蛋白质鉴定信息。而蛋白亚组则更加具体,仅包含共享完全相同肽段集合的蛋白。例如,在采用反奥卡姆剃刀原则时,常通过蛋白亚组划分,以避免形成过于庞大且信息冗余的蛋白组。在宏蛋白质组学研究中,该方法有助于解析单个物种的贡献,即使来自这些物种的蛋白序列高度相似。
蛋白推断方法的选择应与样本复杂性及研究目标相匹配。对于单物种或靶向研究,奥卡姆剃刀原则结合蛋白分组策略可有效减少假阳性鉴定,并简化下游分析。例如,在CAMPI研究中,对扩展型简化人类肠道微生物组(Simplified human intestinal microbiota, SIHUMIx)模拟群落的分析便采用了该策略。而对于复杂的多物种宏蛋白质组样本,通常更倾向于使用反奥卡姆剃刀原则结合蛋白质亚组划分,以最大化蛋白质多样性,同时控制合理的蛋白组规模。例如,在CAMPI研究中,粪便样本分析便采用了这种更具包容性的策略。最终,蛋白推断方法的选择需依据样本的具体特征及研究目标,研究者需在全面的蛋白鉴定与数据的复杂性和可解释性之间做出平衡。
4.1.5 蛋白质定量
蛋白质定量是宏蛋白质组学的核心组成部分,能够提供有关微生物群落功能动态的重要信息。通过蛋白质定量,研究人员可以评估微生物如何响应环境变化,进而揭示生理和代谢过程的变化。例如,营养物质的可用性变化可以导致单一物种或整个微生物群落中蛋白质表达显著改变。本节将概述宏蛋白质组定量中的重要概念、策略和挑战,重点介绍LFQ和有标定量方法,以及下游数据分析的方法。
在宏蛋白质组学的工作流程中,通常采用两种主要的定量策略:LFQ和有标定量。LFQ方法因不需要稳定同位素标记而被广泛使用,尤其适合多样且复杂的样本。常见的LFQ方法包括基于MS1强度的定量和基于MS2谱图计数的定量。MS1定量通过计算每个已鉴定肽段的前体离子强度(如曲线下面积或峰值强度)测量肽段丰度,常用工具包括MaxQuant ,以及独立的工具如moFF或FlashLFQ。与之相比,MS2谱图计数通过匹配的MS2谱图数量对肽段进行定量。尽管MS2谱图计数实施更简单,但其动态范围较窄,精确度略低。目前尚缺乏系统性验证以明确两种方法在宏蛋白质组学中的优劣或适用场景。一项研究表明,谱图计数在合成群落蛋白质生物量评估中比MS1强度更准确。然而,目前领域的共识是,这两种方法通常都适用于宏蛋白质组定量,具体选择取决于研究背景和实验目标。
尽管有标定量方法在蛋白质组学中有广泛应用,但由于微生物群落的复杂性,它们在宏蛋白质组学中使用较少。这些方法,包括TMT和SILAC,能够进行绝对定量,特别适用于需要精确比较样本间差异的实验。然而,对于宏蛋白质组学研究,这些方法实操性受限。环境或临床样本中微生物组的多样性和高复杂性使得标记定量方法不太适用。因此,大多数宏蛋白质组学多采用无标记策略。但有标定量仍适用于目标微生物群落明确的靶向研究。
在宏蛋白质组学研究中,蛋白质定量面临诸多挑战,尤其是在肽段水平将数据整合以推断蛋白质丰度的过程中。该过程受蛋白推断问题的影响,即如何将肽段分配至蛋白质或蛋白质(亚)组(参见第4.1.4节)。大多数软件工具自动完成肽段-蛋白质(组)匹配,从而简化了定量过程。获取蛋白质丰度数据后,需进行数据标准化与转换,以确保统计分析结果的可靠性。尽管已有多种标准化方法被提出用于蛋白质组学数据,但针对宏蛋白质组学的最佳策略仍需进一步探索。
标准化谱丰度因子(Normalized spectral abundance factor, NSAF)是基于谱图计数数据的常用标准化方法,其通过将蛋白质PSM计数除以氨基酸长度校正蛋白大小偏差。随后再通过样本内的总PSM数进行标准化,从而减少批次效应。NSAF计算简便,能够有效应对缺失值问题,尤其适用于宏蛋白质组学的稀疏数据。通常,还会辅以进一步的转换方法,如对数或平方根转换,以满足统计检验的假设要求。
蛋白质组学与宏蛋白质组学的一个关键区别在于,宏蛋白质组学需要考虑微生物群落的多样性和复杂性。在宏蛋白质组学中,可针对特定分类单元进行丰度标准化。为此,已提出基于物种的标准化谱丰度因子(Normalized spectral abundance factor per organism, orgNSAF),这种定向标准化方法使研究人员能够聚焦于特定分类群体内基因表达和功能的变化,从而为微生物活动提供更加细致的见解。
宏蛋白质组数据的独特优势在于可根据科学问题生成多维度数据集。主要整合方式包括:(i)单个蛋白质或同源蛋白质组,这能提供有关群落中单菌特异性功能信息;(ii)生物学功能类别:基于肽段关联蛋白的功能注释,追踪群落功能整体变化;(iii)分类学类别:通过蛋白质丰度估算微生物群落内单个物种的相对贡献。
功能与分类定量的准确性高度依赖数据库注释质量。功能注释可细化至生化反应层级,或宽泛至代谢、基因表达等细胞过程;分类定量分辨率可达菌株/种水平,但当注释不完整时,分类定量可能仅限于较高的分类水平。合理分析后的宏蛋白质组数据能够准确反映群落内物种的蛋白质生物量相对比例,然而,这些定量结果的特异性和准确性与所使用的蛋白质分类注释信息的质量紧密相关。
尽管现有方法可生成有效的数据集,并有助于解析微生物的丰度和功能,但仍需要进一步验证以完善这些方法。目前,宏蛋白质组学中的定量策略仍需要通过基准测试,以确定不同研究类型的最佳方法。未来利用具有成分明确的模拟群落研究,将对宏蛋白质组学中蛋白质定量方法的准确性、可重复性和可靠性进行系统评估。
4.1.6 DIA数据分析
正如第3.7.2节所述,DIA-MS在宏蛋白质组学中的应用需定制化分析流程,以应对微生物群落复杂性与规模带来的独特挑战。与DDA基于筛选高丰度肽段进行碎裂的原理不同,DIA在预设的m/z范围内同时碎裂所有前体离子,从而生成高度复杂的谱图。这种全谱图采集策略需依赖先进的计算工具处理数据。
从DIA-MS中提取定鉴定和定量信息通常需借助专门的软件,如Spectronaut、DIA-NN和EncyclopeDIA。这些工具主要依赖预建谱图库,将实验谱图与理论肽段进行匹配。谱图库通常来源于前期的DDA实验或由蛋白质序列数据库预测生成。尽管无库策略可以直接从蛋白质序列预测谱图,但对复杂宏蛋白质组样本而言,其计算强度过大且缺乏数据缩减策略,难以在实际应用中奏效。为提高鉴定准确性,一种可行的解决方案是利用基因组测序限制数据库检索范围,或通过DDA实验预先构建目标谱图库。尽管这些步骤较为耗费资源,但它们在降低蛋白质和肽段鉴定的模糊性方面至关重要。
宏蛋白质组学数据集的庞大体量进一步放大了DIA-MS分析的挑战,通常涉及数百万条蛋白质和肽段序列。这种数据复杂性带来了巨大的计算需求,并需要完善的数据处理流程。在当前技术条件下,直接对宏蛋白质组学数据集进行无库策略的DIA分析几乎不可行,除非结合基因组测序或基于DDA的光谱库构建。尽管这些准备工作增加了实验复杂性,但却是解析微生物群落动态的关键优化手段。
近年来,质谱技术的进步,如DIA-PASEF 和Orbitrap Astral质谱仪器,显著提升了DIA-MS在宏蛋白质组学中的应用潜力。这些技术能够提升蛋白质组覆盖深度、灵敏度及定量精度。然而,为了充分发挥其优势,需将其与前述的计算工具和谱图库策略紧密结合,以确保其在分析流程中高效合理的应用。
最新基准研究表明,与DDA-MS相比,DIA-MS在宏蛋白质组学分析中的可重复性和准确性更高。利用已知分类组成的模拟微生物群落,DIA-MS在不同实验室间均能稳定鉴定并定量更多肽段和蛋白质,其蛋白质/肽段鉴定重现性更优,且能准确量化蛋白质丰度与分类单元。这些结果进一步凸显了DIA-MS在宏蛋白质组学中的优势,包括深度蛋白质组解析能力、稳健定量性能以及跨样本重现性。然而,当前研究也揭示了DIA工具在处理宏蛋白质组学数据时的不足,亟需提升软件处理微生物组数据的能力。这些发现强调了优化谱图库构建、计算工具及整体分析流程的重要性,以充分展现DIA-MS在微生物群落研究中的潜力。
尽管DIA-MS在可重复性和定量分析方面展现出巨大优势,但其在宏蛋白质组学领域的应用仍处于发展阶段,面临技术与计算双重挑战。随着质谱技术和生物信息学的持续进步,这些难题有望得到解决,从而推动微生物群落功能动态研究的发展。未来仍需不断优化分析流程、改进计算方法,并探索无库策略的可行性,以进一步拓展DIA在宏蛋白质组学研究中的应用。
4.2 分类学和功能分析
在宏蛋白质组学研究中,研究者旨在通过鉴定样本中的微生物种类(分类学分析)并解析其生理功能(功能分析)来表征微生物群落。这些分析有助于深入理解不同环境中微生物群落的组成、多样性及其生态功能。分类与功能注释的准确性依赖于肽段和蛋白质的鉴定质量(见第4.1.1节),并受数据库质量的影响(见第4.1.2节)。本节介绍了宏蛋白质组学研究中用于分类学与功能注释的方法和工具,强调可靠的注释策略及计算资源的重要性。
4.2.1 分类学分析
宏蛋白质组学中的分类学分析通过鉴定样本中表达的蛋白质推断微生物组成,揭示微生物群落结构与多样性,并将蛋白质关联至其分类学来源。分类学注释可通过精确匹配或基于同源性检索实现,常用UniProtKB或NCBI NR等综合数据库。
尽管已有许多专门针对宏蛋白质组学的工具(详见第4.2.4节),研究者亦可使用Centrifuge、Kraken 2等宏基因组学工具。这些工具通过将肽段或蛋白质与已知的分类单元匹配实现注释,但其准确性受公共基因组数据库完整度限制:若样本中微生物未被测序收录,注释可能不完整或不准确。
另一种方法是采用来自宏基因组测序的宏组学数据库。由于蛋白质与基因组紧密相关,将宏基因组序列聚类为MAG可实现基于基因组的分类学注释。例如,GTDB-Tk利用MAG分类信息注释蛋白质分类;对于未关联至MAG的蛋白质,CAT等工具可通过重叠群中所有基因的背景信息推断其分类。随着测序技术的发展,宏基因组组装的精度不断提升,从而进一步提高了物种分类注释的准确性。
4.2.2 功能分析
宏蛋白质组的功能分析揭示微生物群落对环境过程、人类健康与疾病的作用机制。通过量化代谢、运输、复制与防御等功能相关蛋白质丰度,可解析微生物群落的功能动态特征及其在生态系统中的角色。
微生物功能的描述通常依赖于多种功能本体:(1)基因本体(Gene Ontology, GO):将注释分为三类——分子功能、生物过程和细胞成分,分别用于描述基因产物的作用、其参与的生物学过程以及其亚细胞定位;(2)酶学委员会(Enzyme Commission, EC)编号:按催化反应类型对酶进行分类,特别适用于酶活性及代谢通路研究;(3)京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG):将蛋白质映射到代谢和信号通路中,阐释其在生物系统中的作用。
此外,还有一些更为专业的功能本体。例如,MEROPS 专门用于蛋白酶的注释分类,而CAZy 主要针对碳水化合物活性酶(包括糖苷水解酶),它们在分析特定功能类群时提供了更高的特异性。
功能注释通常借助一些常用于宏基因组学的工具,如KoFamKOALA 、InterProScan 和eggNOG-mapper 。尽管这些工具为蛋白质功能注释提供了基本框架,但针对宏蛋白质组学研究的专门工具可提供更精确的注释,相关内容详见第4.2.4节。
4.2.3 基于肽段与基于蛋白的分析方法
在宏蛋白质组学中,分类学和功能分析可以采用基于肽段或基于蛋白质的方法。基于肽段的方法直接依据质谱鉴定得到的肽段,通过匹配已知蛋白质序列的计算机模拟酶切片段,从而进行分类学和功能注释。这种方法能够保留所有潜在来源的蛋白质信息,提供更全面的分类学和功能注释信息。而在基于蛋白质的方法中,首先对应肽段至相应的蛋白质或蛋白(亚)组,通过聚合共享同一蛋白质的肽段来进行分析。这种方法旨在解决蛋白质推断问题,但由于多个蛋白质间存在共享肽段序列,导致肽段归属成为难题。
基于肽段的方法通常会考虑所有潜在来源的蛋白质,而基于蛋白质的方法可能会依据所选的蛋白(亚)组策略剔除冗余信息。这两种方法可能会导致最终注释结果存在差异,关于哪种方法能够提供更准确的结果,目前仍是宏蛋白质组学领域的研究热点。
4.2.4 宏蛋白质组学的分类学与功能分析工具
针对宏蛋白质组学的分类学和功能分析,已有多种工具被开发,并各具特色及适用场景。Unipept是一个强大的工具生态系统,可用于宏蛋白质组学样本的分类学和功能分析,支持命令行界面(Command-line interface, CLI)、桌面应用程序、网页应用及应用程序接口(Application programming interface, API),支持多样化的用户需求与工作流程。该工具采用基于肽段的方法,通过将肽段映射至UniProtKB数据库,直接进行分类学和功能注释。在分类学分析中,Unipept通过计算LCA,即确定所有关联分类中最具体的共享分类层级(图6)。关于LCA计算的详细说明可参阅最新教程。此外,Unipept还支持基于GO、EC编号EC及InterPro分类体系的全面功能分析。对于每个肽段,它会整合所有匹配蛋白质的功能注释并统计频次,结果以表格形式展示于网页端。使用教程与案例见官网(https://unipept.ugent.be/)及已发表文献。
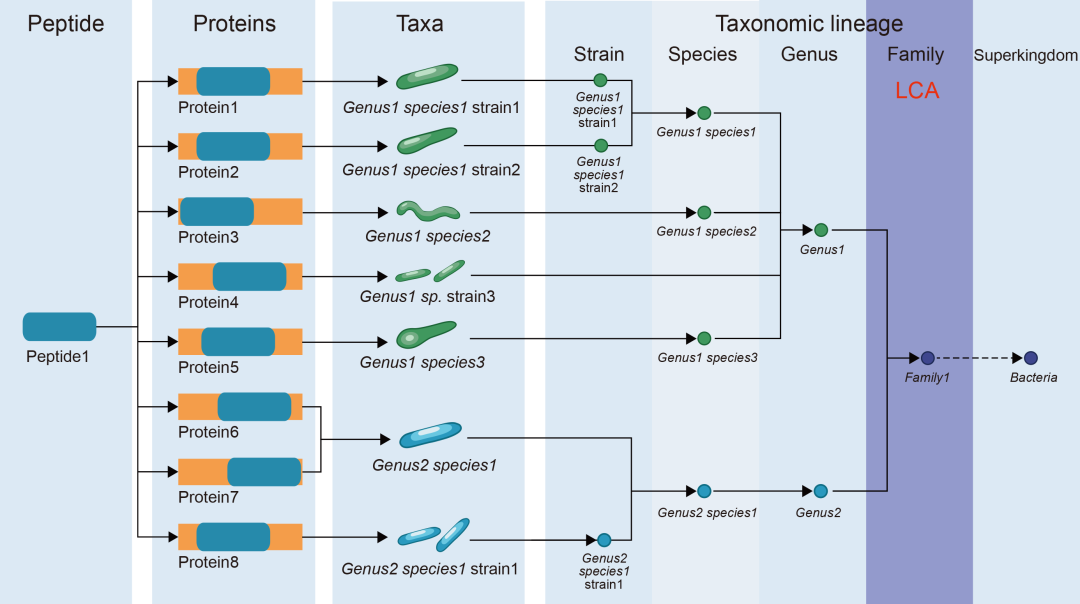
图6. 胰蛋白酶肽段LCA计算示意图
本图假设的肽段1存在于8种不同蛋白质中,对应7个不同物种。通过LCA算法假设其分类学归属为科水平分类单元1。本图改编自[295]。
Peptonizer2000 是一种新型的宏蛋白质组学分类推断流程,能够模拟宏蛋白质组学分析过程中可能引入的误差与不确定性。由于质谱数据分析本身极具挑战,研究人员需要将实验数据与蛋白质序列数据库进行比对,而数据库偏倚、谱图模糊性、冗余肽段序列及跨物种序列同源性等因素均会影响结果。该流程利用贝叶斯统计学将肽段序列、关联分类单元及前期可能的误差建模为概率图谱,随后采用置信传播算法计算概率评分,以推测目标样本中某分类单元的存在可能性。
MetaLab 是一个集成化的软件平台,提供基于质谱原始数据的微生物鉴定、定量与分类分析标准化流程。它采用混合策略,整合基于肽段和基于蛋白质的方法进行宏蛋白质组学分析。MetaLab利用UniProtKB预建索引对肽段进行分类注释,并从eggNOG数据库获取功能注释。最新版本支持DDA和DIA数据分析工作流程,并兼容多种质谱数据格式。有关iMetaLab的详细资源可在其官网查阅(https://wiki.imetalab.ca/)。
Prophane是一款专用于宏蛋白质组学分类学和功能注释的软件,提供交互式可视化结果和直观的网页界面。该工具整合NCBI、UniProtKB、eggNOG、Pfam等多数据库注释数据,全部采用基于蛋白质的方法进行分析。用户可通过Conda软件包(https://anaconda.org/bioconda/prophane)或在线服务(https://prophane.de/login)使用该工具,教程及示例数据集详见官网(https://prophane.de/about/tutorial)。
MetaProteomeAnalyzer(MPA)是一款开源Java工具,支持宏蛋白质组数据的分类与功能分析。MPA结合肽段序列比对和谱图匹配策略,以鉴定样本中的微生物物种与功能通路,使研究人员能够深入探讨微生物群落的代谢活动及其环境互作。该软件兼容多种搜索引擎,并提供基于“宏蛋白”的数据整合功能,以减少冗余数据。桌面端软件及相关教程、文档等资源详见其官网(www.mpa.ovgu.de)。
4.3 下游统计分析
研究人员在分析宏蛋白质组数据时常常面临一个核心挑战:如何选择最合适的下游处理策略。然而,目前尚无统一的、普适性的分析流程可供参考。因此,本节旨在为读者提供构建个性化分析决策树的指导,帮助其根据研究目标与数据特征制定最优方案。在前面的章节中,我们详细介绍了如何生成各种宏蛋白质组数据结果表格,包括肽段、蛋白质、分类学注释以及功能信息。接下来则需通过统计分析挖掘数据中的潜在模式和生物学意义。为了构建稳健的统计分析流程,研究人员需要进行多项关键决策,其要点总结于图7的“速查表”中。
4.3.1 明确科学问题
宏蛋白质组学分析的第一步是明确研究的核心科学问题。宏蛋白质组学能够进行多种研究,以下是一些常见的研究问题示例(图7A):(一)队列研究:健康与疾病个体的差异特征是什么?是否存在特定疾病的潜在生物标志物?(二)微生物组动态功能变化:微生物群在时间和空间上如何变化?功能生态层面是否呈现β多样性?特定环境因素对微生物组有何影响?(三)扰动研究:微生物群落如何在分类学、功能和生态学层面上响应外界干扰?(四)多组学研究:通过整合宏蛋白质组学与其他组学方法,能获得哪些系统性见解?
4.3.2 选择分析层级
在明确研究问题后,下一步是选择适当的分析层级,以便有效解答这些问题(图 7B)。不同的研究目标需要不同的分析方法,主要包括以下三种:
i)基于特征的分析
基于特征的分析方法是宏蛋白质组学中最常见的分析策略,主要聚焦于识别差异特征,即在不同组或实验条件下表现出显著性差异的可量化变量。例如,特定肽段、蛋白质、分类单元或功能类别的丰度变化都可以作为研究对象。
在基于特征的分析中,有两个关键原则需要考虑:(i) 统计分布假设:通常假设数据符合标准统计分布(如正态分布),以便选择合适的分析方法。(ii) 特征独立性假设:将特征视为相互独立的变量,使得参数检验和非参数检验等传统统计方法可适用。遵循这些原则,基于特征的分析能够有效识别跨数据集的生物学差异特征。
ii)基于群落的分析
与基于特征的分析不同,基于群落的分析将整个数据集视为一个动态的生态群落,而不是独立的个体特征。在这一框架下,蛋白质被看作是互作网络组成部分,其功能通过进化关系和分类学起源紧密联系。例如,不同分类单元的蛋白质可能表现出功能冗余,而生态动态则可能影响功能与分类群的相互作用。
由于群落内存在复杂的相互作用,传统的统计方法(通常假设特征独立性)可能并不适用。因此,宏蛋白质组学借鉴宏基因组学的发展开发了新生态学分析方法。
例如,功能冗余度量:利用二分网络关联分类和功能属性,以评估群落的健康状态和稳定性。同样,PhyloFunc方法:通过在系统发育树的节点上整合功能距离,并采用类似UniFrac的加权策略,进行功能β多样性分析。该方法能够区分功能变化的来源——究竟是近缘物种之间的补偿效应,还是远缘物种间的类群转换,为生态动态机制研究提供了重要见解。
iii)跨组学分析
宏蛋白质组学与其他宏组学(如宏基因组学、宏转录组学)存在内在关联,因此,整合多组学数据对于深入理解微生物群的系统生态学至关重要。不同的组学方法各具优势,它们共同反映了分子生物学中心法则(DNA → RNA → 蛋白质)的不同层级的动态变化,有助于全面解析微生物群的生物学过程和生态相互作用。
尽管这些组学数据能够互补,传统研究通常仍然采用各自独立的工作流程进行分析。然而,近年来,随着生物信息学工具和平台(如Galaxy 和 MOSCA )的发展,促进了跨组学整合分析。这种整合方法不仅提升了数据解析深度,也有助于更深入地理解群落生态的功能动态。
在一项最新研究中,研究人员结合宏基因组学和宏蛋白质组学,通过比较群落内基因组与蛋白质组层面的功能冗余差异,评估特定蛋白质是发挥生态位功能(支撑微生物群落在环境中的生态角色)还是执行必需代谢功能。基因-蛋白丰度差异较大可能表明某些基因虽然存在,但未表达蛋白质,暗示它们具有更专一化的生态位功能。而基因-蛋白丰度差异较小则可能表明这些基因被积极转录并翻译为蛋白质,说明它们在群落中执行核心代谢功能。

图7. 宏蛋白质组学下游数据分析“速查表”
(A)宏蛋白质组学下游分析关注的核心问题领域。(B)确定研究目标层级,以便选择合适的分析策略。(C)合理选择数据预处理流程。(D)选择适当的数据分析方法集合。
4.3.3 数据预处理方法
在确定合适的分析层级后,下游数据分析的首要步骤是数据预处理。常见的数据预处理步骤包括数据过滤、数据转换、数据填补和数据标准化(图7C)。然而,并不存在普适性数据预处理方法,最佳数据预处理策略需依据具体的研究问题来选择。
i)数据转换
在蛋白质组学和宏蛋白质组学研究中,常用的数据转换方法包括对数转换(如log₂或log₁₀)和平方根转换。但并非所有场景均适用于数据转换。
数据转换适用场景:当需使数据接近正态分布时,建议进行数据转换。在特征层面的分析中,对峰强度进行对数转换可使数据更近似正态分布,而许多常见的宏蛋白质组学特征选择方法(如线性模型、经验贝叶斯、单变量 t 检验、偏最小二乘判别分析(Partial least squares discriminant analysis, PLS-DA)和正交偏最小二乘判别分析(Orthogonal partial least squares discriminant analysis, OPLS-DA))均依赖正态分布假设。如果数据偏离正态分布,可考虑采用非参数检验方法。
数据转换不适用的场景:需反映蛋白质真实丰度时,应避免数据转换。例如,火山图常用于识别差异特征,其x轴表示对倍数变化,y轴表示统计显著性(- log₁₀(p-value))。尽管数据经对数转换便于可视化,但在统计分析或比较时应保留原始未转换数据。此外,在群落层面的分析中,对数转换可能会掩盖蛋白质生物量信息,而生物量信息对于估算分类和功能组成至关重要。蛋白质强度或PSM计数可作为分类单元生物量贡献的可靠指标。因此,在基于组成的分析(如 α 和 β 多样性或功能冗余度评估)中,建议使用未转换数据。
ii)数据中心化与标准化
标准宏蛋白质组学研究流程中,为确保数据的一致性和可比性,通常会从每个样本中提取等量的蛋白质,进行酶解,并加载至质谱仪器。然而,在特定研究场景下,宏蛋白质组学可能基于固定体积体系的总蛋白质生物量而非标准化蛋白质含量进行定量。在这种情况下,不建议进行数据中心化和标准化,而应采用总谱图计数标准化或中位数标准化等替代方法。
iii)数据过滤
数据过滤通常用于去除噪声、无关特征或异常值,其应用需结合研究背景定制。
对于特征层面的分析,严格的数据过滤至关重要,尤其是在鉴定生物标志物时。通常要求蛋白质在大多数样本中均被检测到,以确保所鉴定生物标志物的稳定性。例如,研究者可设定较高的检出率阈值(如要求蛋白质在70%–90%样本中存在)。这种筛选有助于降低假阳性率,增强生物标志物的可信度。此外,其他类型的特征层面分析同样需要严格的数据过滤。然而,数据过滤的阈值及方法(如针对整个数据集或按组进行过滤)需谨慎选择,避免过度过滤导致剔除真正有生物学意义的特征。
相比特征层面的分析,群落层面的数据过滤更为宽松,以便全面反映群落动态特征。适度的过滤有助于去除明显噪声,但一般不会设置过于严格的阈值。例如,未过滤的分类特异性功能数据有利于完整展示微生物组功能分布。
iv)数据填补
在宏蛋白质组学数据集中,缺失值通常由两种机制导致:宏蛋白质组本身多样性高且稀疏性显著,导致大量真实缺失蛋白质(非随机缺失);现有宏蛋白质组学技术的检测深度有限,导致低丰度蛋白可能难以在所有样本中被检测到(随机缺失)。
数据填补需极度谨慎。不当的填补方法可能会导致假阳性结果。当某个特征缺失数据比例高(如>50%),过度填补可能引入虚假值,导致假阳性结果。此外,如果数据填补方法未能准确反映缺失数据的真实机制,则可能引入系统性偏倚,尤其当数据同时存在非随机缺失和随机缺失时。如果数据在填补后被用于统计检验,应返回原始未填补数据二次验证差异真实性,以确保结论的可靠性。
另一种替代方案是采用单变量选择方法,该方法联合检验缺失性与类别关联性及观测强度差异,这种方法可作为数据填补的替代方案。
需要注意的是,数据填补对特征选择分析至关重要,而在群落层面分析中通常是不必要的,其原因与前述内容相同。
4.3.4 数据分析方法的选择
在深入理解和谨慎选择数据预处理步骤后,下游数据分析的最后一步是选择合适的分析方法(图7D)。此阶段为数据挖掘提供了重要契机,研究人员可以通过多种策略深入挖掘数据,揭示有意义的生物学或生态学模式与结论。这一过程通常是最具探索性且耗时的部分,常用策略包括但不限于以下几类:
降维分析:降维方法常用于揭示数据集中的潜在模式或结构,并评估样本间的相似性。无监督方法,如PCA、t分布随机邻域嵌入(t-distributed Stochastic Neighbor Embedding, t-SNE)、层次聚类和k均值聚类广泛应用。此外,PLS-DA等有监督方法也经常使用。降维分析不仅适用于肽段、蛋白质、物种分类和功能数据,还可用于MS1谱图数据,尤其当研究重点是揭示样本间模式时。
富集分析:富集分析用于评估某一特征子集在背景数据库中是否显著过表达,以识别可能具有生物学意义的功能模块。虽然可以使用R等编程语言实现富集分析,但 iMetaShiny 提供了交互式功能,支持蛋白质ID或直系同源群簇(Clusters of Orthologous Groups, COG)ID 的分类学和功能富集分析。然而,目前基于蛋白质ID的富集分析仅适用于基于人体肠道整合基因目录(Integrated Gene Catalog, IGC)数据库研究。
特征选择:已开发多种在线工具(如MetaFS、MetaQuantome 、MetaX 和iMetaShiny )以及独立运行的软件(如Meta4P ),以简化宏蛋白质组数据的特征选择分析。这些工具无需深入的编程知识,使研究人员能够高效地提取关键生物信息。
通路分析:通路分析通常用于概览检测到的功能,或比较组间的差异表达和富集通路。KEGG mapper 和iPath 是最为常用的通路分析工具。新开发的PathwayPilot进一步优化了KEGG通路分析流程,该工具利用EC编号识别活性酶,作为KEGG代谢通路中相关代谢物,便于解析特定条件下生物功能特征,并对支持目标物种靶向分析。
群落分析:除了基于特征的分析,群落水平的分析将整个宏蛋白质组视为一个动态生态系统,重点研究其整体功能和组成特征。这类分析可能涉及群落组成推断、α多样性、β多样性及功能冗余度评估,为理解微生物群落的功能动态提供更全面的视角。
5. 共同合作:携手宏蛋白质组学倡议成员撰写全面综述
宏蛋白质组学倡议是一个致力于推动微生物组研究中宏蛋白质组学领域发展的国际性社区。在人类蛋白质组织(Human Proteome Organization, HUPO)和欧洲蛋白质组学协会(European Proteomics Association, EuPA)的支持下,与欧洲生命科学基础设施ELIXIR合作,该倡议作为核心平台,促进研究者交流技术进展、共享研究方法并建立宏蛋白质组学领域的标准。
该倡议旨在促进专家与入门研究者之间的交流,规范实践方法,并加速宏蛋白质组学方法学的发展。其核心使命是成为宏蛋白质组学基础理论、前沿进展和应用研究的权威资源库,通过构建协作网络推动实验和生物信息学方法的发展进步。
宏蛋白质组学倡议的三大支柱为:(1)沟通与合作:聚焦领域进展分享,组织如CAMPI等基准研究,并举办国际宏蛋白质组学研讨会(International Metaproteomics Symposium, IMS);(2)教育与推广:通过网络研讨会、培训课程等易获取资源,向更多的微生物组研究人员普及宏蛋白质组学知识,并促进专家之间的交流;(3)标准化:致力于制定健全的(元)数据标准,推行FAIR数据原则,以确保研究成果的开放性与可复用性。
作为我们在教育与推广方面承诺的一部分,我们编写了本篇综述,以向读者普及宏蛋白质组学。为了确保内容全面且视角均衡,我们首先邀请各领域专家起草撰写独立章节。随后进行内部审阅,并通过初期反馈优化各章节内容。作者完成修订后,文稿经历多轮交叉审阅,使所有贡献成员都能共享见解并处理遗留问题。我们也邀请了宏蛋白质组学领域的入门研究者对文稿进行审阅,以确保内容对该领域以外的研究人员清晰易懂。在整合他们的反馈后,所有合著者(包括各章节作者以及专家和新手审阅者)共同进行了终稿审定。这种协作模式使我们得以完成一部既深入全面又易于理解的文稿资源,并在正式投稿前以预印本的形式公开分享。
6. 结 论
《微生物学研究者的宏蛋白质组学指南》旨在为初入宏蛋白质组学领域的微生物组研究者提供实践性入门指导,降低技术壁垒。本指南涵盖宏蛋白质组学核心内容,包括实验设计、样本制备、质谱数据采集、肽段鉴定、蛋白质推断、分类和功能分析,以及基础统计方法。它为微生物学和微生物组研究提供了应用宏蛋白质组学技术所需的基本知识。宏蛋白质组学是一个快速发展的领域,仍然存在诸多技术挑战与未探索领域。本指南侧重于基础概念,而非对该领域进行详尽概述。为此,宏蛋白质组学倡议发起“CAMPI”系列研究,旨在推动多实验室协作,对样本制备、质谱分析方法和生物信息学流程进行比对和优化。展望未来,随着质谱技术的持续革新,宏蛋白质组学分析的覆盖深度将进一步提升。结合实验方案、研究策略及生物信息学工具的持续改进,将共同推动该领域的发展。以CAMPI为代表的协作模式,充分体现推动宏蛋白质组学发展中的协作力量。这些研究进展,加之微生物组研究人员的贡献,将帮助我们更深入地理解各类生态系统中微生物组及其功能的认知。
数据可用性声明:
本文未生成或使用需公开的数据集。补充材料(图文摘要、幻灯片、视频、中文版翻译及更新材料)可通过在线DOI或 iMeta Science网页http://www.imeta.science/ 获取。
引文格式:
Tim Van Den Bossche, Jean Armengaud, Dirk Benndorf, Jose Alfredo Blakeley-Ruiz, Madita Brauer, Kai Cheng, et al. 2025. “The Microbiologist’s Guide to Metaproteomics.” iMeta e70031. https://doi.org/10.1002/imt2.70031.
作者简介

Tim Van Den Bossche博士(第一作者)
● 比利时根特大学(Ghent University)VIB-UGent博士后研究员。
● 聚焦于开发和应用前沿的计算方法,以解析复杂微生物群体中蛋白质的表达与功能,特别关注在宏蛋白质组数据的处理、功能注释和生态信息挖掘研究。国际宏蛋白质组学倡议管理员。

李乐园(通讯作者)
● 国家蛋白质科学中心(北京)特聘研究员;
● 研究方向为宏蛋白质组与微生物系统生态学。主持国家自然科学基金面上项目等科研项目4项。以第一/通讯作者在iMeta、Nature Communications、Microbiome、Gut Microbes、npj Biofilms and microbiomes等期刊发表学术论文20余篇。Journal of Proteomics 执行编辑,国际宏蛋白质组学倡议国际通讯员。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,中科院分区生物学1区Top,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 5872
5872

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









