






点击蓝字 关注我们
微生物组单细菌RNA测序揭示人类肠道关键菌种未被探索的特化代谢功能

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接: https://onlinelibrary.wiley.com/doi/full/10.1002/imt2.70035
● DOI: https://doi.org/10.1002/imt2.70035
● 2025年4月17日,浙江大学王永成/沈一飞等在iMeta在线发表了题为“Single-microbe RNA sequencing uncovers unexplored specialized metabolic functions of keystone species in the human gut”的文章。
● 本研究通过对三种不同肠型的健康供体应用新型的肠道微生物组单细菌RNA测序及分析框架,构建了人类肠道微生物组的转录图谱,并在单细菌层面上分析了肠道菌群的代谢能力的异质性和协变关系,解析了亚洲人群的关键菌种——单形巨单胞菌的未被探索的特化代谢功能。
● 第一作者:沈一飞、瞿闻馨、宋孟迪、张天宇
● 通讯作者:王永成(yongcheng@zju.edu.cn)、沈一飞(yifeishen@zju.edu.cn)、郑书发(zsfzheng@zju.edu.cn)
● 合作作者:刘畅、施晓锋、徐欣欣、姜晶晶、丁利国、莫钫宇、茅浙英、黄明珠、徐子叶、陈嘉业、沈恩惠、阮健、刘炯、Michael P. Timko、陈瑜、樊龙江
● 主要单位:浙江大学医学院附属第一医院、良渚实验室、M20 Genomics、浙江大学生物信息所、美国弗吉尼亚大学
亮 点
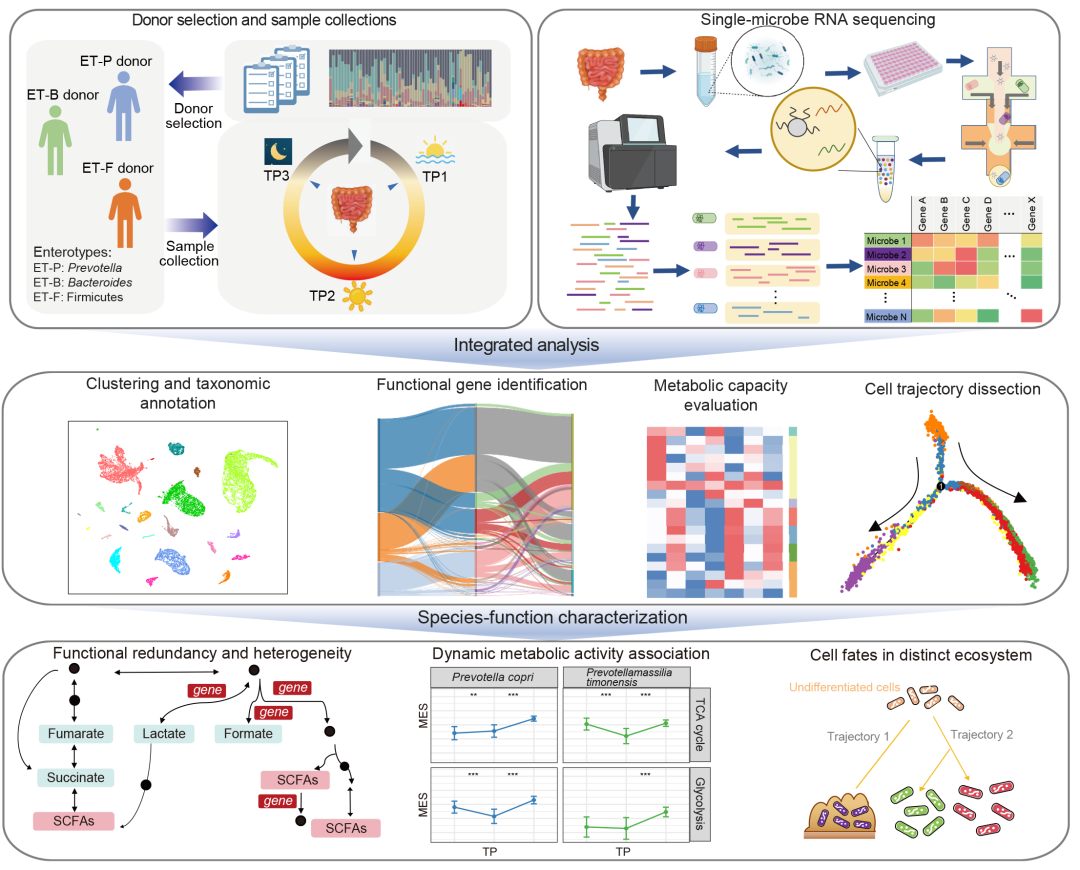
● 本研究通过对三种不同肠型的健康供体应用新型的肠道微生物组单细菌RNA测序及分析框架,构建了人类肠道微生物组的转录图谱;
● 研究了肠道菌群在中心碳代谢中的功能冗余与互补性,并在单细菌层面上分析其代谢能力的异质性和协变关系;
● 亚洲人群的关键菌种——单形巨单胞菌可通过降解外源性植酸提高人体矿物质吸收率。
摘 要
人体内栖息着数万亿微生物,这些微生物在健康和疾病中发挥着关键作用。尽管我们对人类肠道微生物组的菌种和功能组成的认识正在迅速扩展,但目前认知主要仍基于丰度测量。因此,目前对人体微生态系统中的菌种—功能异质性和动态活动仍知之甚少。通过应用新型肠道微生物组特异性单细菌RNA测序技术及分析框架,本研究对三种不同肠型的健康供体进行研究,绘制了包含198个菌种、38,922个细菌的肠道微生物组转录图谱,并深入解析其功能。重点研究了短链脂肪酸相关中心碳代谢中的功能冗余与互补性,并分析了单细菌代谢能力的异质性和协同变化。通过比较一天中不同时间点的肠道微生物组,我们对肠道菌群的昼夜动态活动与功能异质性的关系进行探索。值得注意的是,通过单细菌RNA测序技术,我们系统解析了亚洲人群关键菌种——单形巨单胞菌(Megamonas funiformis)的代谢功能异质性。结合体外和体内实验验证,证实了该菌能通过降解外源性植酸有效提高人体矿物质吸收率,这一发现使其有望成为改善矿物质缺乏所致营养不良的潜在益生菌。我们的研究结果表明:菌种—功能异质性在人类肠道微生物组中广泛存在并具有重要作用,而单细菌RNA测序技术能有效捕获转录活性差异,从而鉴定出具有重要生物学和临床价值的特化代谢功能关键菌种。
视频解读
Bilibili:https://www.bilibili.com/video/BV1uBGtz5EzY/
Youtube:https://youtu.be/dOYbDvzfros
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
人类胃肠道由数量庞大、种类繁多的微生物组成,这些微生物与健康和多种疾病状态密切相关。肠道菌群在宿主的代谢过程中发挥着关键作用,包括分解复杂碳水化合物和蛋白质、产生必需微量营养素和短链脂肪酸(SCFAs)。菌群内部的功能受到微生物竞争、生态位以及宿主驱动的共生选择和功能冗余压力的影响。然而,细菌分离培养的技术限制阻碍了对肠道微生物的功能表征。
肠型分析能有效划分人群,并提供肠道微生物组成个体差异信息。人类肠道中存在数百种细菌,其中厚壁菌门和拟杆菌门是优势菌门。目前,我们对其功能异质性、生态系统冗余和互补模式的理解仍然有限。在揭示菌种—功能活性异质性和冗余性方面仍存在重大挑战。
肠道菌群在调节昼夜节律中的作用对于理解多种生理过程具有重要意义。肠道菌群组成的昼夜节律对代谢功能至关重要,然而,目前对肠道菌群与宿主昼夜节律相互作用的理解主要基于微生物相对丰度的变化,而对肠道微生物组的功能昼夜动态活动的认识甚少。要充分理解这些相互作用的生态和进化意义,需要借助新技术设计微生物组研究。
单细菌RNA测序技术的最新进展为研究复杂微生物群落提供了新视角,能以单细菌分辨率评估菌群功能异质性、生态位以及对宿主和环境因素的适应性响应。在本研究中,我们结合肠道特异性单细菌RNA测序方法和转录分析框架:(1)构建人类肠道微生物组的全面转录图谱,并解析肠道菌种的功能特征;(2)探究短链脂肪酸相关中心碳代谢中的功能冗余与互补性;(3)研究单细菌代谢能力的异质性和协变关系;(4)探索微生物组在肠道生态系统多样性中的昼夜动态活动的功能变化;(5)探索不同生态位中细菌状态转变,并鉴定人类肠道微生物组中关键菌种的特化代谢功能。
结 果
人类肠道微生物组单细菌RNA测序的研究设计概述
针对不同肠型特征的研究揭示了以核心指示菌群为中心的微生物共现网络:拟杆菌肠型(ET-B)以拟杆菌属(Bacteroides)为标志菌,普雷沃氏菌肠型(ET-P)由普雷沃氏菌属(Prevotella)主导,而厚壁菌肠型(ET-F)则由厚壁菌门(Firmicutes)主导。
为系统研究人类肠道微生物组的转录活性,我们基于内部队列的宏基因组分析,纳入了三位具有代表性肠型的健康供体(ET-P、ET-B、ET-F)。ET-F供体中最优势的菌种是厚壁菌门成员——单形巨单胞菌Megamonas funiformis。值得注意的是,在欧洲和美洲人群研究中,巨单胞菌属从未被报道为优势菌属,但在中国和日本人群中被普遍检出,提示其可能是亚洲人群的特征性菌属。尽管已有研究证实单形巨单胞菌与人类健康相关,但其在肠道微生物组中的具体功能机制仍不清楚。为探究肠道微生物组的昼夜动态变化,我们在一天内三个时间点(清晨:TP1;正午:TP2;夜间:TP3)采集了每位供体的粪便样本(图1A),所有样本均在24小时内完成采集。通过单细菌RNA测序(图1B)和整合数据分析(图1C),我们揭示了肠道微生物组的菌种-功能异质性及昼夜动态活动规律(图1D),并进行了实验验证(图1E)。
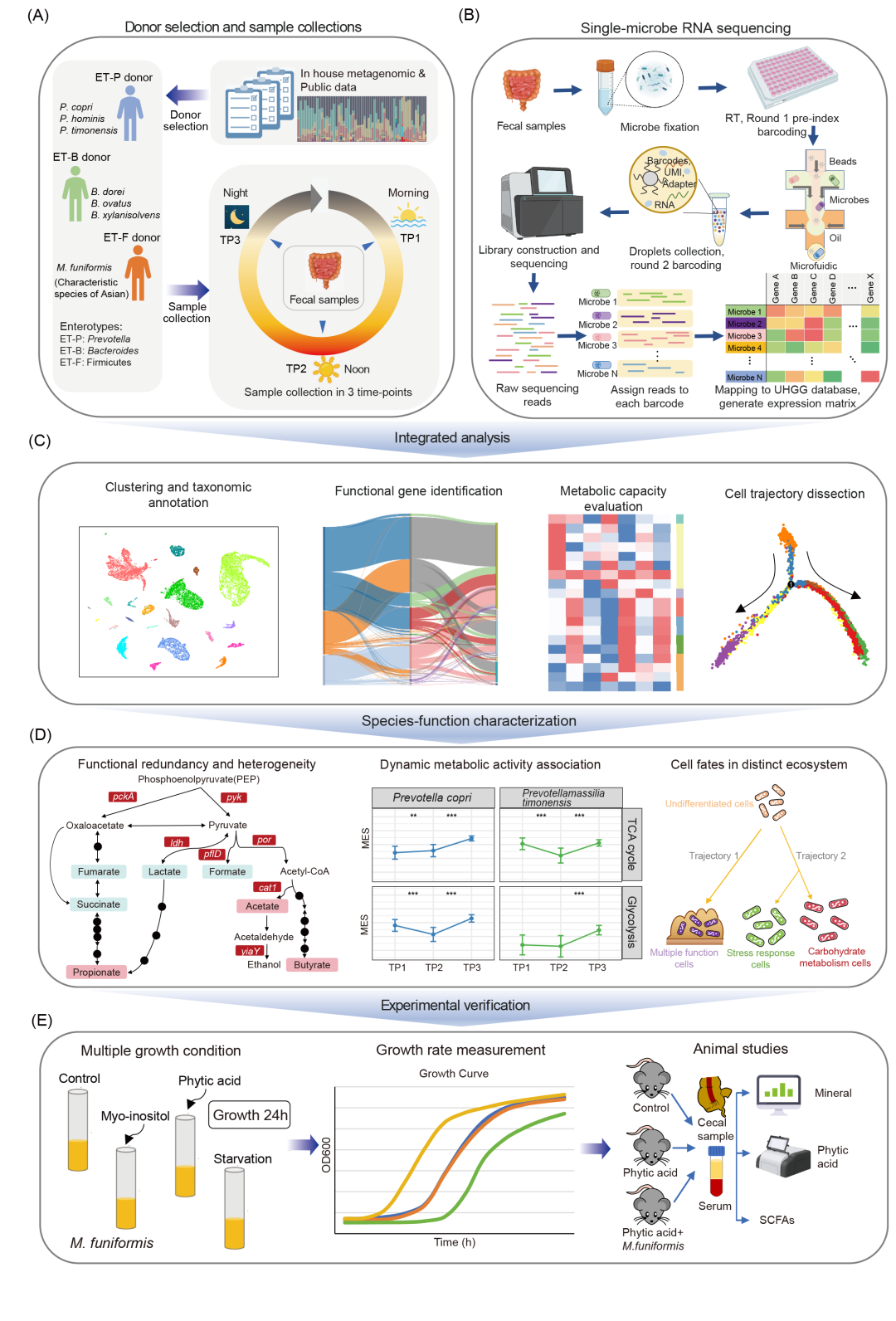
图1. 人类肠道微生物组单细菌RNA测序实验设计概览
(A)供体选择与样本采集方案;(B)单细菌RNA测序流程;(C)数据分析框架;(D)菌种—功能异质性、昼夜动态及细菌状态转变特征;(E)关键功能菌种(M. funiformis)实验验证。
单细菌转录图谱揭示人类肠道菌群的菌种特异性功能特征
人类肠道微生物组包含大量种类繁多的微生物,且不同个体间菌种组成差异显著。采用之前开发的微生物组注释流程,我们在三位供体中共鉴定出198个菌种,并筛选出30个核心菌种(每个菌种包含超过100个细菌)进行后续功能分析。随后,我们将单细菌分类注释与UMAP降维聚类结果整合(图2A)。不同聚类簇明确对应特定菌种,其中M. funiformis、P. copri和B. dorei是三位供体中最优势的菌种(图2B)。
利用各菌种对应的转录聚类簇,我们通过鉴定菌种特异性标志物基因进一步分析功能特征(图2C)。在三位供体共有的16个菌种中,共鉴定出220个功能标志物基因,其中37个(16.8%)与氨基酸代谢相关,15个(6.8%)涉及能量代谢,6个(2.7%)参与脂肪酸代谢(图2D)。值得注意的是,101个(45.9%)标记基因与碳水化合物代谢相关,这些基因在M. funiformis、P. copri、P. timonensis和B. dorei中显著富集,表明肠道菌种在碳代谢中存在功能异质性。
功能标记基因分析还揭示了与硫代谢、细菌运动和应激反应相关的菌种特异性特征。例如:人类结肠中常见的硫酸盐还原菌D. piger显著表达参与异化亚硫酸盐还原的aprA和dsrB基因(图2E);健康人群常见的毛螺菌科成员CAG-81属高表达与细菌运动和宿主免疫应答相关的鞭毛蛋白基因hag(图2F),这与既往研究认为毛螺菌科是肠道主要鞭毛蛋白产生者的结论一致。此外,我们在拟杆菌属菌种中观察到超氧化物歧化酶基因(sodB)的高表达(图2G)。总之,单细菌转录图谱揭示了菌种特异性功能特征,有助于发现人类肠道微生物组中具有特定功能的菌种。
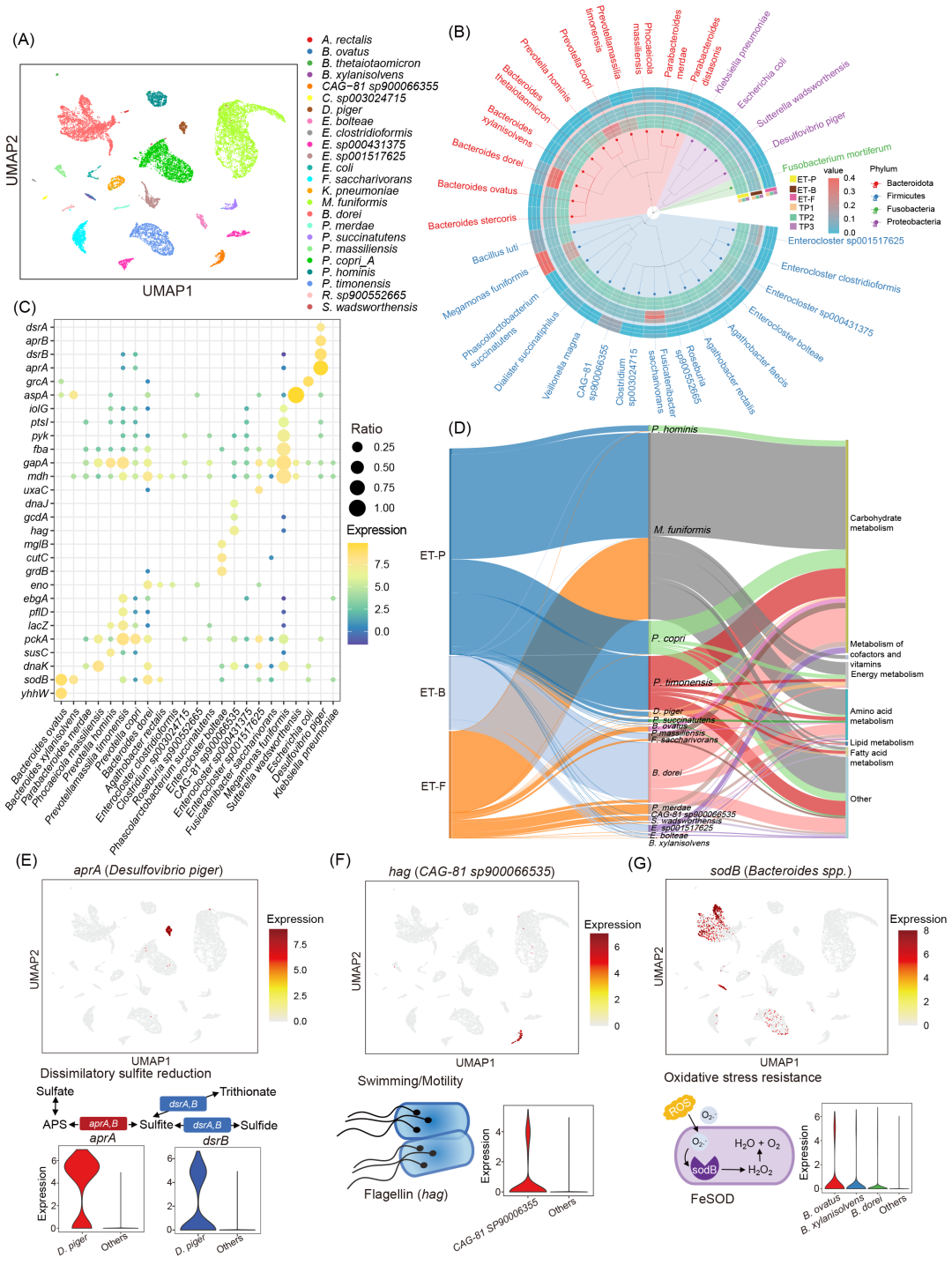
图2. 人类肠道单细菌转录图谱及菌种特异性功能特征
(A)基于菌种注释的UMAP降维图;(B)九个样本微生物分类组成系统树;(C)各菌种功能标志基因点图;(D)菌种标志基因与功能的桑基图;(E)硫代谢相关基因aprA表达量的UMAP图及异化亚硫酸盐还原通路的示意图;(F)鞭毛蛋白基因hag表达量的UMAP图;(G)超氧化物歧化酶基因sodB表达量的UMAP图。
短链脂肪酸相关中心碳代谢的功能冗余与互补性分析
鉴于菌种特异性功能标志物基因中碳代谢相关通路基因的显著富集,接下来,本研究对各菌种的中心碳代谢相关基因进行了深入解析(图3A)。在初级降解中,我们观察到ET-P供体的普雷沃氏菌属中淀粉和蔗糖代谢相关基因(如glgP、pulA和amyA)的表达量达到峰值(图3B)。已知普雷沃氏菌富含多种植物多糖代谢酶。编码β-半乳糖苷酶的lacZ基因在ET-P的7个菌种中均有表达,其中P. timonensis表达水平最高。半乳糖代谢相关基因(如galM和galK)同样在ET-P供体的普雷沃氏菌属中最高表达。
在初级发酵中,磷酸己糖变位酶家族基因pgm在ET-P的P. timonensis和ET-B的B. dorei中高表达(图3B)。糖酵解途径相关基因在各供体优势菌种中表达最高(图3C)。对维持碳稳态及提供核苷酸和氨基酸合成前体至关重要的磷酸戊糖途径(PPP)也呈现显著基因表达特征(图3D):编码葡萄糖-6-磷酸脱氢酶(zwf)和6-磷酸葡萄糖酸脱氢酶(gnd)的基因在ET-B的B. dorei和A. rectalis中高表达。进一步分析发现,非氧化性PPP核心转化反应相关基因——转酮醇酶(tktA)和转醛醇酶(talB)在ET-P供体的P. timonensis和ET-B供体的B. dorei中高表达。在次级发酵中,PEP羧激酶(pckA)和丙酮酸激酶(pyk)分别催化PEP向草酰乙酸和丙酮酸的转化。pckA在ET-P的P. timonensis和ET-B的E. sp001517625中表达最高,而pyk在ET-P和ET-F的M. funiformis中表达最显著(图3C)。作为TCA循环关键组分的草酰乙酸,其合成相关基因在ET-B拟杆菌属中表达最高(图3E)。
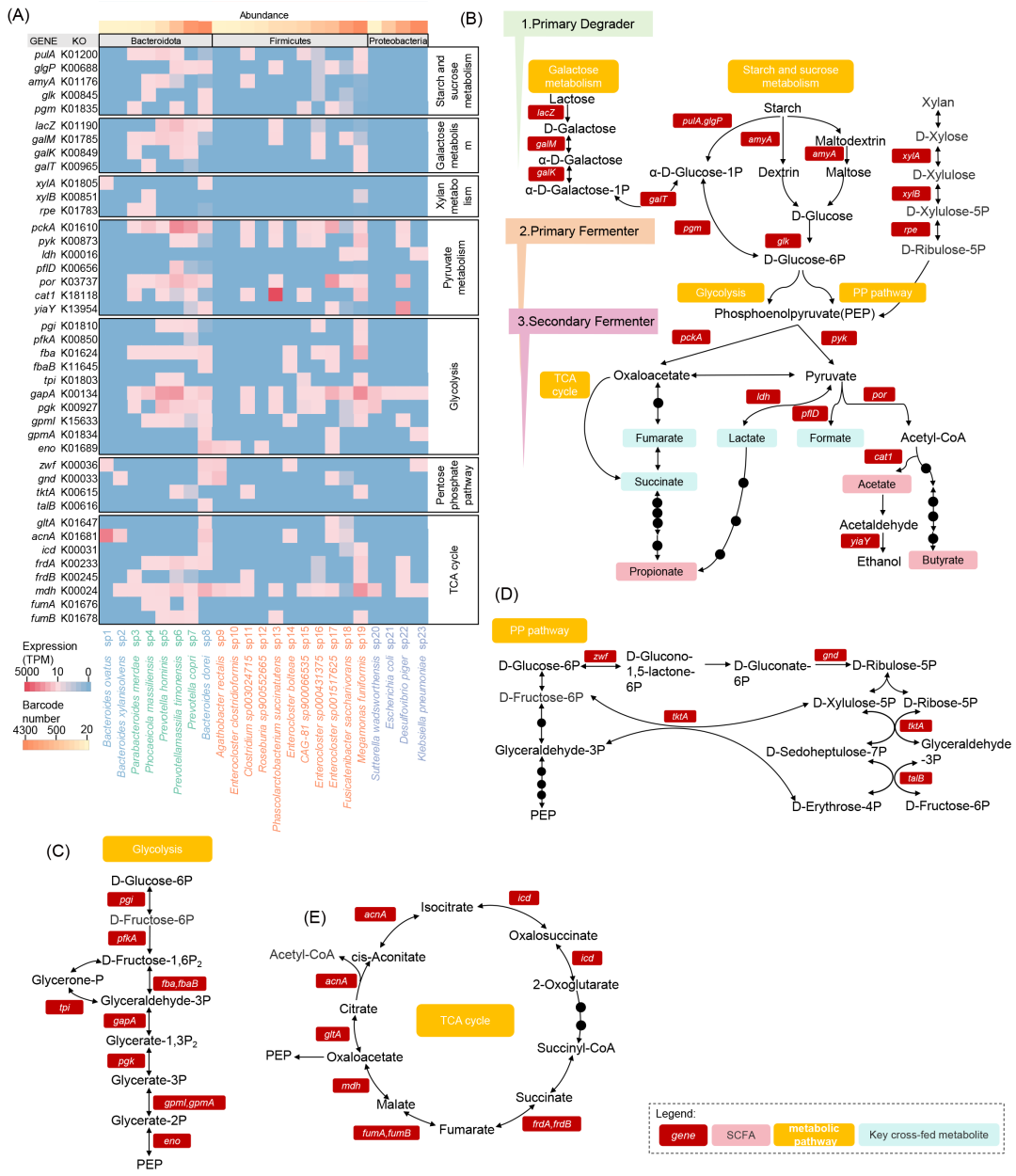
图3. 短链脂肪酸相关中心碳代谢的功能冗余与互补性
(A)碳水化合物代谢基因表达热图;(B)半乳糖、淀粉、蔗糖、丙酮酸代谢通路;(C)糖酵解通路;(D)磷酸戊糖途径;(E)TCA循环。图示肠道微生物三级营养代谢:初级降解菌(第一营养级)水解多糖,初级发酵菌(第二营养级)生成PEP,次级发酵菌(第三营养级)产生SCFAs。
人类肠道菌群代谢功能的异质性与协同性
基于单细菌分析观察到的肠道菌群代谢相关基因变化,为系统解析其代谢能力异质性,本研究对各菌种进行了单细菌代谢基因富集分析(图4A)。,并开发了微生物-代谢分析流程,MIC-Metabolism(图4B)。MIC-Metabolism包含三个步骤:(1)代谢基因功能注释;(2)基于排序的代谢富集评分(MES)生成;(3)通过置换检验实现MES标准化及菌种活性推断。应用该流程分析三位供体的单细菌RNA测序数据,评估了各菌种的代谢通路活性(图4C,D)。
结果发现,在ET-P的普雷沃菌属及M. funiformis中,糖酵解和丙酮酸代谢通路的代谢富集得分MES最高(图4D),这与图3结果一致,表明这些菌种主要参与肠道碳代谢。D. piger的硫代谢通路显著激活,与图2E发现相符。ET-F的M. funiformis中,TCA循环、糖酵解和丙酮酸代谢通路MES最高(图4E),而B. dorei的丙酸代谢通路活性最高。ET-B中,B. dorei的丙酸代谢通路MES最显著(图4F)。
针对B. dorei和M. funiformis在不同肠型供体中的表现,我们对二者进行了跨供体代谢能力比较。与ET-B相比,ET-F的糖酵解、丙酮酸代谢和TCA循环通路活性显著降低(图4G),但ET-P在细菌分泌系统、蛋白质外运和精氨酸合成通路的MES显著高于ET-F(图4H),揭示了肠道菌种在不同微环境中的代谢差异。
通过MES相关性分析,发现ET-P中P. timonensis与M. funiformis在碳代谢中显著共激活。ET-F的B. dorei与E. sp000431375呈显著正相关(图4I,J)。
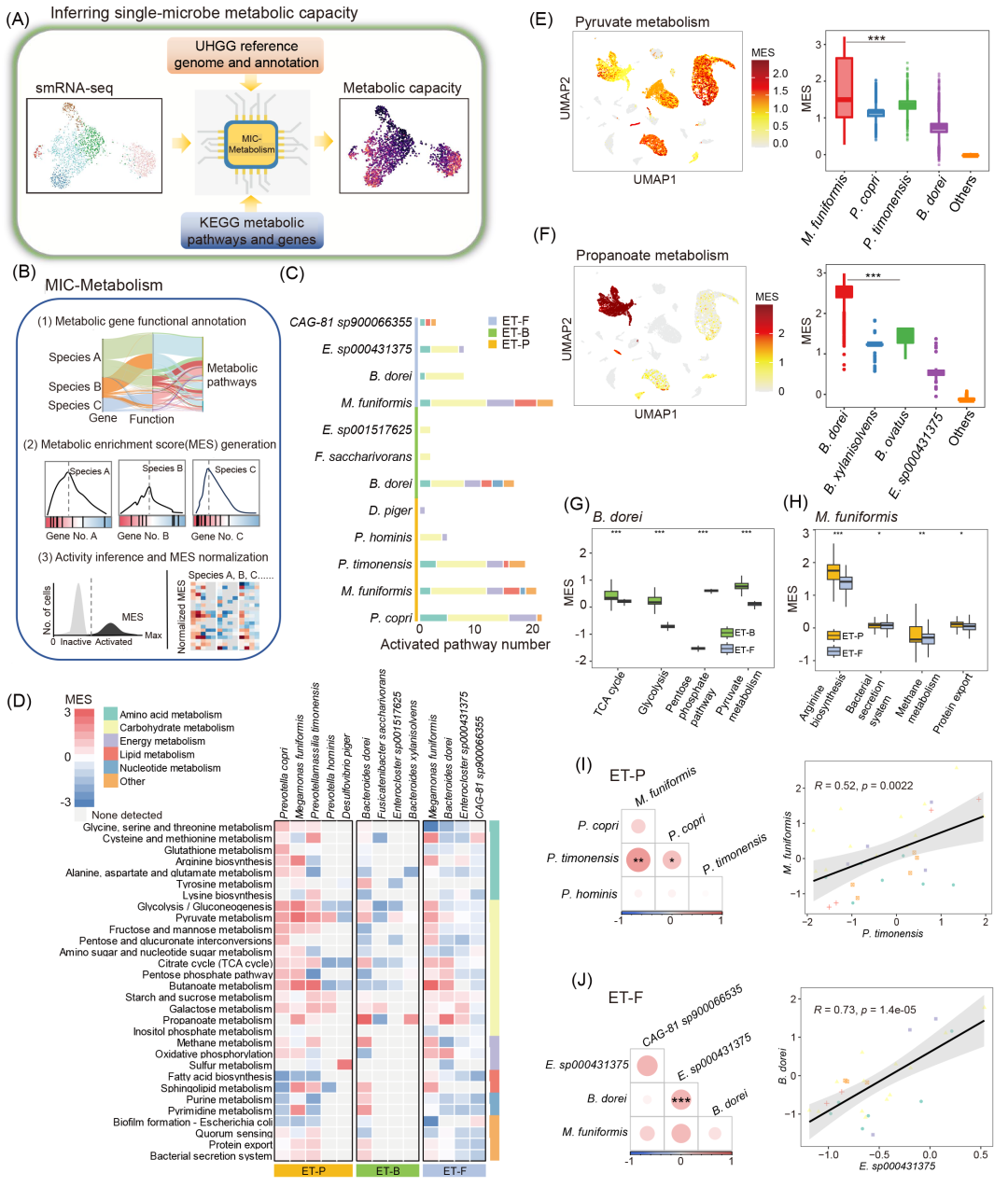
图4. 单细菌代谢功能异质性及协变关系
(A)MIC-Metabolism简介;(B)MIC-Metabolism分析流程概览;(C)三个供体内部分菌种激活的代谢通路数量;(D)代谢通路MES得分热图;(E)丙酮酸代谢的MES得分用UMAP展示(左)和MES比较(右);(F)丙酸代谢的MES用UMAP展示(左)和MES比较(右); (G)B. dorei跨供体间的MES差异;(H)M. funiformis跨供体间的MES差异;(I)ET-P供体部分菌种的MES相关性;(J)ET-F供体部分菌种MES相关性。*p<0.05,**p<0.01,***p<0.001。MES:代谢富集得分。
肠道菌群的功能异质性相关的昼夜动态活动
为在单细菌分辨率下全面解析人类肠道微生物组的功能动态,我们对三名健康供体一天内三个时间点的样本进行了深入分析。对单细菌RNA测序数据的无监督聚类分析显示,部分菌种在不同时间点存在显著转录差异(图5A)。通过鉴定图示菌种的时间特异性标志基因发现,三个供体中TP2时间点的标志基因数量较少,而ET-P供体TP3时间点的标志基因数量增多(图5B)。基因功能分析显示,多数时间特异性标志基因与碳水化合物代谢和氨基酸代谢相关(图5C)。
通过MIC-metabolism计算各时间点的代谢通路MES,发现不同菌种的代谢活动呈现显著异质性(图5D)。例如ET-P中,P. copri、P. timonensis和M. funiformis的TCA循环、糖酵解及丙酮酸代谢通路MES呈现相似动态模式,而ET-B仅B. dorei在这些通路中表现动态变化。MES相关性分析显示:ET-P中丙酮酸代谢与糖酵解通路在P. copri、P. timonensis和M. funiformis间协同激活(图5E)。这些结果揭示了肠道菌群动态互作的复杂性,其可能受昼夜节律、饮食和微生态位等因素调控。
基于观察到的细菌动态活动,我们推测昼夜波动可能与亚群功能异质性相关。通过对ET-B和ET-P的优势菌种(B. dorei和P. copri)进行深入分析发现:P. copri的三个亚群中,TP3富集的亚群(簇1)代谢活性显著更高,其中果糖二磷酸醛缩酶基因(fba)和PEP羧激酶基因(pckA)表达上调(图5F)。ET-B的B. dorei可分为五个功能亚群:氧化磷酸化细胞(簇0)、mutA+丙酸代谢细胞(簇2)和aspA+丙氨酸/天冬氨酸/谷氨酸代谢细胞(簇3)。这些功能亚群在不同时间点呈现特异性富集:氧化磷酸化细胞主要存在于TP3,氨基酸代谢细胞富集于TP1,而丙酸代谢细胞在TP2占优(图5G)。

图5. 单细菌RNA测序捕获肠道微生物组昼夜动态
(A)UMAP图分别按照菌种以及时间点着色;(B)热图展示时间点标志基因的数量;(C)时间标志基因功能桑基图;(D)TCA循环、糖酵解、丙酮酸代谢的MES随时间变化的连线图;(E)ET-P内的部分菌种代谢通路相关性网络;(F)P. copri亚群分析;(G)B. dorei亚群分析。
单形巨单胞菌在不同生态位中的功能状态分析
M. funiformis是亚洲人群肠道微生物组的特征性菌种。尽管其健康重要性已被认知,但该菌在复杂肠道菌群中的代谢模式与功能仍不清楚。通过MIC-metabolism分析,我们发现不同供体肠道菌群中的M. funiformis存在代谢差异(图3)。为深入解析其功能异质性,我们从单细胞转录组中提取该菌进行分群与功能解析。UMAP聚类分析将所有M. funiformis划分为7个功能亚群(图6A)。其中簇0、2、6主要来自ET-F,簇1、3、5源于ET-P,簇4则在两供体中均存在(图6A)。通过标志物基因鉴定与代谢能力评估发现:ET-F主导的簇2高表达蛋白质折叠相关基因(如clpB及伴侣蛋白dnaK、htpG);ET-P的簇3显著表达精氨酸合成、肌醇磷酸代谢、蛋白质外运和生物膜形成相关基因(图6B)。MES比较显示:簇0在中心碳代谢通路(TCA循环、糖酵解和丙酮酸代谢)活性最高(图6C),簇3的肌醇磷酸代谢和蛋白质外运通路MES显著高于其他亚群(图6D,E)。
拟时序分析发现三个明确的细胞命运轨迹(图6F):未分化细胞(主轨迹,951个细胞)、多功能细胞(命运1,869个细胞)和应激细胞(命运2,2403个细胞)。多功能细胞特异性表达蛋白质转运酶亚基(secY、secA)、精氨酸合成(argB-D、argF-G、argJ)及肌醇磷酸代谢(iolB、iolD-E、iolG、iolI)相关基因,应激细胞则高表达伴侣蛋白基因。细胞代谢命运沿未分化细胞→多功能细胞→应激细胞方向演变。拟时序分析鉴定出269个高变基因(图6G)。Wilcoxon秩和检验证实多功能细胞在"大肠杆菌生物膜形成"通路中活性显著高于应激细胞。富集基因主要集中于“糖原生物合成”通路——该过程通过细菌产生胞外多糖并转运至细胞外环境或膜结构促进生物膜发育,在细胞状态转换中起核心作用(图6H)。值得注意的是,应激细胞的代谢活性存在显著异质性:碳水化合物代谢细胞的中心碳代谢活性明显高于应激响应细胞。这些发现为单细菌分辨率下解析肠道微生态系统的功能异质性提供了新视角。
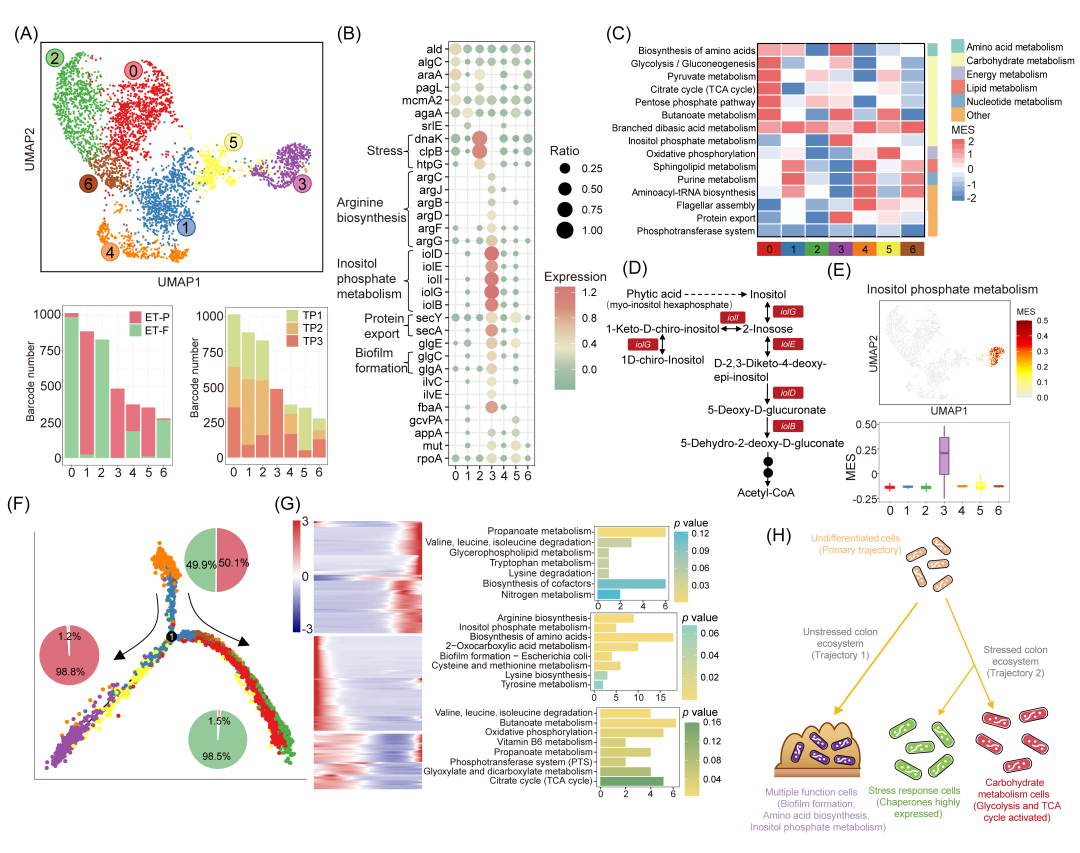
图6. M. funiformis细菌状态转换的功能轨迹分析
(A)七亚群UMAP聚类;(B)亚群标志物基因点图;(C)通路MES热图;(D)肌醇磷酸代谢通路;(E)亚群MES比较;(F)拟时序分析;(G)拟时序高变基因;(H)生态位中细菌状态转换。
多组学整合分析与分子枢纽机制解析单形巨单胞菌通过降解外源性植酸促进矿物质吸收
基于M. funiformis应激基因和肌醇相关基因的高表达以及肌醇磷酸代谢通路的激活,我们设计了一系列实验探究其潜在机制。实验设置了三种培养条件:高浓度肌醇、高浓度植酸(肌醇六磷酸,IP6)以及营养缺失环境。从哥伦比亚血琼脂(CBA)平板上选取单菌落后,首先测定不同条件下M. funiformis的生长曲线,进而在分子水平量化基因表达差异;随后检测植酸处理组的植酸浓度以评估其降解能力;最后,通过测定培养上清液中短链脂肪酸(SCFAs)含量分析植酸降解产物(图7A)。
生长曲线显示:植酸组中M. funiformis生长更快且iolD基因表达更高,而添加肌醇对生长速率影响甚微(图7B,C),提示肌醇相关基因的高表达可能源于该菌对植酸的代谢。营养缺失条件下细菌生长显著受限,同时应激相关基因显著上调(图7C)。生长稳定期培养上清液中的植酸浓度较对数期显著降低(图7D)。SCFAs检测发现:植酸组生长稳定期的培养上清液的乙酸和丙酸水平显著高于对数期,而丁酸无显著变化(图7E),说明该菌能将植酸降解为乙酸和丙酸。为探究M. funiformis在肠道菌群中的功能,我们开展了动物实验。7周龄C57BL/6J小鼠经一周适应后随机分为三组(对照组、植酸组、植酸+M. funiformis组),连续灌胃两周。鉴于高植酸饮食会抑制矿物质吸收,我们检测了血清矿物质浓度,并通过测定血清植酸和回盲部的SCFAs含量评估M. funiformis的植酸降解能力(图7F)。
结果显示:植酸组血清钙(Ca)、镁(Mg)、锌(Zn)水平显著降低,而M. funiformis添加组显著高于植酸组(图7G),与既往研究一致。M. funiformis添加组血清植酸水平显著低于植酸组(图7H),说明该菌在体内具有植酸降解能力,此外,M. funiformis添加组的回盲部乙酸和丙酸水平显著高于对照组与植酸组,而丁酸无显著变化(图7I),与体外实验结果吻合。这些证据表明M. funiformis能有效降解植酸并生成乙酸和丙酸,有望成为改善高植酸饮食所致矿物质缺乏的潜在益生菌(图7J)。
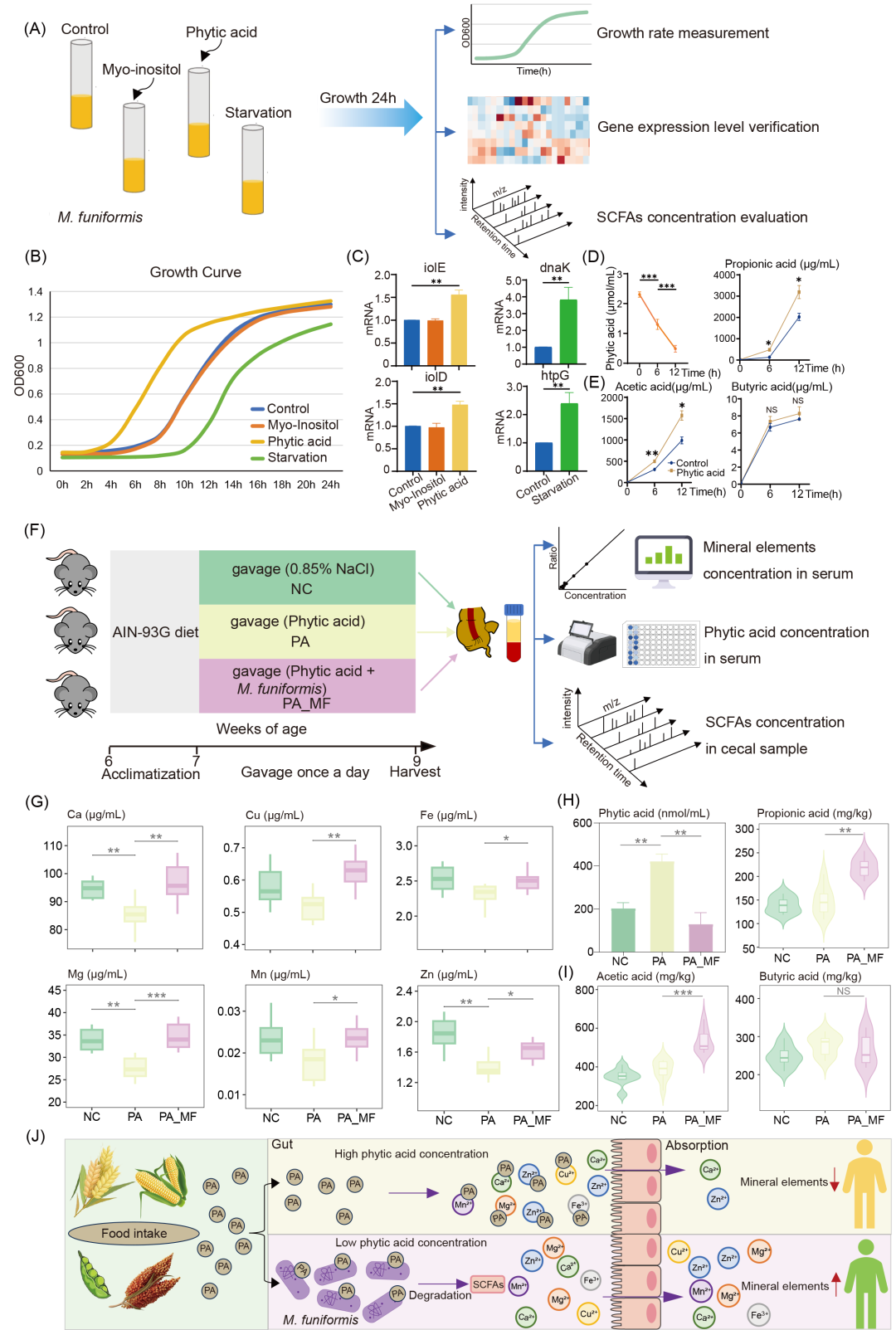
图7. M. funiformis通过降解外源植酸促进矿物质吸收
(A)体外实验设计概览;(B)M. funiformis在不同培养条件下的生长曲线;(C)M. funiformis在不同培养条件下的关键基因表达;(D)不同培养条件下培养基上清液植酸浓度变化;(E)不同培养条件下培养基上清液SCFAs的量;(F)体内实验设计概览(n=8);(G)不同组别小鼠血清矿物质含量水平;(H)不同组别小鼠血清植酸浓度;(I)不同组别小鼠回盲部SCFAs含量;(J)M. funiformis通过降解外源植酸促进矿物质吸收概览。NC:生理盐水组;PA:植酸组;PA_MF:植酸+菌剂组。NS:无显著性。Cu:铜;Fe:铁;Zn:锌;Mn:锰。
讨 论
本研究采用单细菌RNA测序技术,在单细菌的分辨率上,解析了三种不同的肠型(ET-P、ET-B和ET-F)健康供体肠道微生物组的功能冗余和异质性。通过分析三位供体一天内三个时间点的共计九份样本,我们揭示了菌种间与菌种内的功能异质性特征。前期研究通过高通量单细菌RNA测序已成功量化肠道微生物的菌种间与菌种内适应策略异质性,本研究在此基础上重点探究了不同肠型人群肠道菌群的特异功能异质性和动态活动变化。
现有研究已证实人类肠道中不同菌种存在功能的分化。我们的结果表明,基于单细菌转录特征的功能多样性可被有效捕获。针对跨供体共有的菌种,我们进一步发现同一菌种的代谢功能会随宿主环境改变而调整。例如M. funiformis在ET-P中氨基酸代谢更活跃,而在ET-F中脂代谢活性更高(图4D),这提示细菌代谢功能可能受饮食或宿主遗传背景等因素调控。功能冗余是肠道微生物组的普遍现象,我们通过单细菌RNA测序全面解析了其模式:在ET-P中,拟杆菌门的P. copri与厚壁菌门的M. funiformis均表现出高碳水化合物代谢活性(图3A,4D)。这种功能相似菌种的共存具有生态学意义,当某一有益菌缺失时,其他菌种可提供补偿性功能。微生物功能多样性及冗余机制对维持生态系统稳定性至关重要。
本研究首次在单细菌分辨率下捕获了肠道微生物组的昼夜动态活动。既往关于饮食干预影响菌群的研究多关注菌种丰度组成变化,传统宏基因组技术仅能提供菌种相对丰度信息。然而微生物群落的行为和生物学效应不仅取决于菌种组成,更与每个微生物的细菌状态密切相关。通过分析一天内三个时间点的粪便样本,我们获得了新的认知:例如ET-F中M. funiformis的菌量虽无显著波动,但在单细菌水平其碳水化合物代谢活性呈现显著动态变化,这可能是对进食或宿主生理节律的响应。
特别值得注意的是,我们在亚洲人群特征菌种M. funiformis中鉴定出具有不同代谢特征的亚群。这些亚群在代谢、应激响应等通路的基因表达存在显著差异,其中部分亚群显著激活肌醇磷酸分解代谢通路,提示其可能参与植酸(肌醇六磷酸,IP6)降解。植酸作为植物性饮食中的主要抗营养因子,在谷物、坚果等亚洲主食中含量较高。本研究发现植酸能显著促进M. funiformis生长,动物实验证实该菌可通过提高钙、镁、锌的生物利用率作为益生菌发挥功能。相较于传统的菌特化代谢功能的筛选方法,本研究为探索关键功能菌种提供了新思路。
本研究存在以下局限:样本量较小(每种肠型仅一位供体),后续需扩大样本量并设置餐前对照;实验设计上仅采用植酸代谢菌(大肠杆菌K-12)作为阳性对照而未设阴性对照,矿物质检测仅针对血清样本,且使用的M. funiformis菌株非直接分离自特定供体。这些局限为未来研究指明了改进方向。
结 论
综上所述,本研究通过单细菌RNA测序技术,以前所未有的高分辨率系统解析了人类肠道微生物组的三大核心特征:菌种特化功能与冗余性、昼夜动态活动规律及微生物状态转换异质性。单细菌RNA测序技术能更精准地揭示不同生态位中菌群的动态转变过程,显著深化了对个体间"菌种-功能"异质性的认知。此外,本研究所开发的单细菌RNA测序与分析框架,为鉴定肠道微生物组中具有重要生物学和临床价值的关键功能菌种提供了高效策略。新发现单形巨单胞菌通过降解植酸促进矿物质吸收的独特功能。这些创新性发现与方法学突破,将为精准微生物组诊断和靶向菌群干预提供全新工具,最终助力人类健康水平的提升。
方 法
粪便样本采集
本研究从三名不同肠型的健康男性供体(20-30岁)采集九份粪便样本,采样时间点为:早餐后(TP1,6-9时)、午餐后(TP2,11-13时)和晚餐后(TP3,18-21时)。供体保持日常饮食习惯。样本经4℃、500g离心3分钟去除食物残渣和宿主细胞后,进行宏基因组测序以确定肠型分类(属水平)。纯化样本再经4℃、3900g离心5分钟收集菌体用于单细菌RNA测序。
肠道微生物组单细菌RNA测序
微生物固定与透化
粪便样本直接重悬于4%多聚甲醛中,4℃过夜固定。200g离心5分钟后,上清经细胞筛过滤,用含RNase抑制剂的PBS洗涤,最后重悬于含0.04% Tween-20的PBS中。菌体与细胞壁消化酶37℃孵育15分钟后,用PBS-RI终止反应并重悬于DEPC水(约500万细菌/27.5μL)。
原位反转录与dA加尾
使用VITApilote-PFT1200试剂盒(M20 Genomics),菌体与反转录体系(含随机引物、dNTPs和逆转录酶)经8-42℃梯度退火后30℃延伸。反转录产物用PBS-T洗涤三次后,加入TdT酶体系37℃反应30分钟进行dA加尾。
单细菌液滴包裹
按已发表的流程,将菌体与DNA延伸反应混合液、条形码水凝胶微球注入微流控芯片,生成单细菌液滴后经37℃(1h)-75℃(20min)梯度孵育。
cDNA富集
液滴经全氟辛醇处理后,用磁珠纯化。qPCR确定最佳循环数后,进行PCR扩增,产物经Qubit 3.0定量和片段分析仪质检。
文库构建与测序
使用Vazyme DNA文库构建试剂盒,cDNA经末端修复、腺苷化后连接接头,PCR扩增文库。质检合格后在NovaSeq 6000平台进行150bp双端测序。
数据分析
数据质控与注释
通过MIC-Anno流程过滤低质量条形码并注释菌种。保留含>100条形码的核心菌种进行分析。使用STAR(v2.7.10a)将reads比对至UHGG(v2.0.1)参考基因组,featureCounts(v2.0.3)和umi_tools(v1.1.2)定量基因表达。
降维聚类与功能标记基因鉴定
表达矩阵导入Seurat(v4.1.3),保留nCount_RNA>30且nFeature_RNA>15的微生物。PCA降维后通过UMAP聚类(分辨率0.5),使用Wilcoxon秩和检验(校正p<0.05)鉴定亚群标记基因。ANOSIM分析时间点差异。
MIC-Metabolism分析
基于KEGG通路基因集,采用ssGSEA算法计算代谢富集评分(MES)。1000次置换检验(p<0.01为显著激活)和z-score标准化后,通过Spearman相关性分析菌种间通路共激活模式。
拟时序分析
使用Monocle 2(v2.26.0)分析M. funiformis状态转换。q<0.01的差异基因用于构建DDRTree轨迹,clusterProfiler(v4.6.0)进行KEGG富集分析。
生长曲线实验
从德国DSMZ获取M. funiformis DSM 19343。CBA平板厌氧培养后,挑取单菌落接种于CMC液体培养基。OD600达0.6时,分别接种于含50μg/mL肌醇、植酸或70%营养缺失培养基中(n=5),每2小时测OD600吸光度值。收集植酸组0/6/12h培养上清测植酸和SCFAs。阳性对照选用大肠杆菌K-12(ATCC 25404)。
RT-qPCR分析
提取RNA并反转录,CFX96系统(Applied Biosystems)进行SYBR Green qPCR。16S rRNA归一化后ANOVA统计分析。
动物实验
取6周龄C57BL/6J小鼠随机分为三组(n=8/组):植酸组(0.2mg/g体重)、生理盐水对照组、植酸+M. funiformis组(1×109 CFU/只),连续灌胃2周。第3周禁食12小时后采集血清(测矿物质/植酸)和盲肠内容物(测SCFAs)。
血清矿物质检测
血清经硝酸微波消解后,ICP-MS(PerkinElmer,美国)检测Cu、Fe、Zn、Mn、Mg、Ca浓度。
植酸检测
使用Solarbio试剂盒,血清或培养上清经提取液I/II处理后,测定OD700值并计算植酸浓度。
SCFAs检测
回盲部内容物样本或培养上清经研磨、酸化和甲基叔丁基醚萃取后,GC-MS(Agilent,美国)分析。以2-乙基丁酸为内标。
统计分析
Wilcoxon秩和检验(Bonferroni校正)分析微生物转录差异,ANOSIM检验时间点差异,ANOVA比较基因表达,Spearman相关分析通路共激活。
代码和数据可用性
本研究产生的单细菌RNA测序数据集已存储于基因组序列归档库(Genome Sequence Archive),生物项目编号为“PRJCA036556”(访问链接:https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA036556)。实验源数据已随本文提供。MIC-Metabolism分析流程代码及单菌RNA测序数据的深度分析方法已发布于Github平台(https://github.com/MIC-seq/MIC-seq-analysis-workflow/tree/main/MIC-Metabolism)。补充材料(包括附图、附表、图文摘要、演示文稿、视频、中文翻译版本及更新资料)可通过在线DOI或iMeta Science官网获取(http://www.imeta.science/)。
引文格式:
Yifei Shen, Wenxin Qu, Mengdi Song, Tianyu Zhang, Chang Liu, Xiaofeng Shi, Xinxin Xu, Jingjing Jiang, Liguo Ding, Fangyu Mo, Zheying Mao, Mingzhu Huang, Ziye Xu, Jiaye Chen, Enhui Shen, Jian Ruan, Jiong Liu, Michael P. Timko, Yu Chen, Longjiang Fan, Shufa Zheng, Yongcheng Wang. 2025. “Single-microbe RNA sequencing uncovers unexplored specialized metabolic functions of keystone species in the human gut.” iMeta 4: e70035. https://doi.org/10.1002/imt2.70035.
作者简介

沈一飞(第一/通讯作者)
●浙江大学医学院附属第一医院检验科,全省临床体外诊断技术研究重点实验室PI。
●研究方向为微生物组单细胞测序及多组学多模态人工智能诊断。近三年在iMeta、Protein & Cell、Advanced Science、Gut Microbes、Clinical Chemistry、Briefings in Bioinformatics等期刊发表SCI论文多篇。

瞿闻馨(第一作者)
● 浙江大学医学院附属第一医院。
● 研究方向为多组学多模态人工智能诊断。以共同一作在iMeta、Gut Microbes等期刊发表SCI论文4篇。
宋孟迪(第一作者)
●浙江大学良渚实验室。
●研究方向为单细胞转录组在微生物中应用。

张天宇(第一作者)
●M20 Genomics。
●研究方向为基于随机引物的单细胞测序数据分析,以第一作者(含共同)在iMeta、Nature Communications、Angewandte Chemie、Protein & Cell等期刊发表SCI论文4篇。

郑书发(通讯作者)
●浙江大学医学院附属第一医院特聘研究员,副教授。
●研究方向为感染性疾病的实验诊断研究,浙江省杰出青年科学基金获得者,入选浙江省卫生高层次人才和浙江大学临床拔尖人才。主持国家自然科学基金2项,国家重点研发计划课题1项,以第一或通讯作者(含共同)在BMJ、Advanced Science等期刊发表SCI论文30余篇。以主要完成人获国家科技进步奖特等奖、中华医学科技奖一等奖、教育部技术发明二等奖各1项。

王永成(通讯作者)
●浙江大学良渚实验室、浙江大学医学院附属第一医院“百人计划”研究员,博士生导师。
●研究方向为微流控和单细胞分析技术开发。入选国家级高层次人才、杭州市顶尖人才、《麻省理工科技评论》中国“35岁以下科技创新35人”等,主持中国空间站项目、国家自然科学基金、浙江省领军型创新团队、浙江省尖兵研发攻关项目等纵向项目及多项企业横向项目。共发表50多篇论文,共被引用5000多次,近3年以通讯作者在Nature Microbiology、Nature Protocols、Nature Reviews Genetics、Nature Communications等期刊发表近20篇论文。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,中科院分区生物学1区Top,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,目标是成为影响因子大于15的高水平综合期刊,欢迎投稿!
"iMetaMed" 是“iMeta” 子刊,专注于医学、健康和生物技术领域,目标是成为影响因子大于15的医学综合类期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
出版社iMetaMed主页:
https://onlinelibrary.wiley.com/journal/3066988x
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
iMetaMed投稿:
https://wiley.atyponrex.com/submission/dashboard?siteName=IMM3
邮箱:
office@imeta.science

















 2697
2697

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









