






原文GWAS分析数据质控
GWAS分析数据质控
1、格式转换
第一种常用的格式:plink格式
- 正常格式
map和ped:比如a.ped,a.map - 二进制文件
bim,bed,fam:比如a.bed, a.bim, a.fam
第二种常用的格式:vcf格式
第三种常用的格式:hapmap格式
1.1 plink正常格式转二进制格式
比如这里有plink格式的文件,前缀为a的plink文件:
$ ls
a.map a.ped
将其转化为二进制文件:b.bed, b.bim, b.fam
plink --file a --out b
结果:
$ ls b*
b.bed b.bim b.fam b.log
注意:
- 如果染色体超过23,比如30对染色体,需要设定--chr-set 30
- 如果有非数字染色体,比如性染色体,需要设定--allow-extra-chr
- 常用的动物都有对应的参数,直接设定相关动物就行,比如牛的--cow,下面是其它动植物的。如果没有对应的物种,直接设置染色体的条数以及允许非数字染色体即可。
--cow
--dog
--horse
--mouse·
--rice
--sheep
1.2 plink二进制格式转为正常格式(map和ped)
这里有plink格式的文件,前缀为b的plink二进制文件:
$ ls b*
b.bed b.bim b.fam b.log
将其转化文件:c.map, c.ped
plink --bfile b --recode --out c
注意:
- –bfile,因为输入文件b*为二进制,所以用–bfile,如果是一般格式,用–file即可
- –recode,要输出正常格式,所以用–recode指定,如果不加这个参数,默认是输出二进制文件
- –out,输出文件的前缀
结果:
$ ls *c*
c.hh c.log c.map c.ped
1.3 正常plink文件转为vcf文件
这里有plink格式的文件,前缀为c的plink二进制文件:
$ ls *c*
c.hh c.log c.map c.ped
将其转化文件:d.vcf
plink --file c --recode vcf --out d
注意:
- –file,用–file指定正常plink格式的文件
- –recode vcf,要输出vcf文件格式
- –out,输出文件的前缀
文件预览:
1.4 二进制plink文件转为vcf文件
和正常plink文件类似,除了--file 变为--bfile即可。
现有文件:
$ ls e.vcf
e.vcf
plink --vcf e.vcf --recode --out f
注意:
- –vcf 需要文件名完整,不能只写前缀,所以这里要写
--vcf e.vcf - –recode 保存plink文件
保存为二进制文件:
plink --vcf e.vcf --out g
结果:
$ ls g*
g.bed g.bim g.fam g.log
2、常用质控
2.1、SNP缺失质控。
输入文件前缀:HapMap_3_r3_1(HapMap_3_r3_1.bed HapMap_3_r3_1.bim HapMap_3_r3_1.fam)
#查每个个体和每个 SNP 的缺失情况
plink --bfile HapMap_3_r3_1 --missing
输出:plink.imiss 和 plink.lmiss,这些文件分别显示了每个个体缺失 SNP 的比例和每个 SNP 缺失个体的比例。
#根据缺失情况绘制直方图
Rscript --no-save hist_miss.R
hist_miss.R 脚本内容:
indmiss<-read.table(file="plink.imiss", header=TRUE) #每个个体缺失 SNP 的比例
snpmiss<-read.table(file="plink.lmiss", header=TRUE) #每个 SNP 缺失个体的比例
# read data into R
pdf("histimiss.pdf") #indicates pdf format and gives title to file
hist(indmiss[,6],main="Histogram individual missingness") #selects column 6, names header of file
pdf("histlmiss.pdf")
hist(snpmiss[,5],main="Histogram SNP missingness")
dev.off() # shuts down the current device
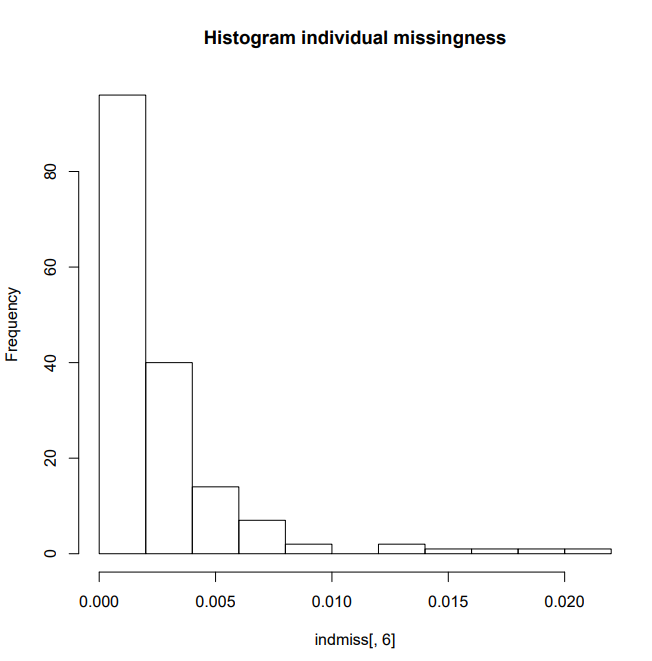
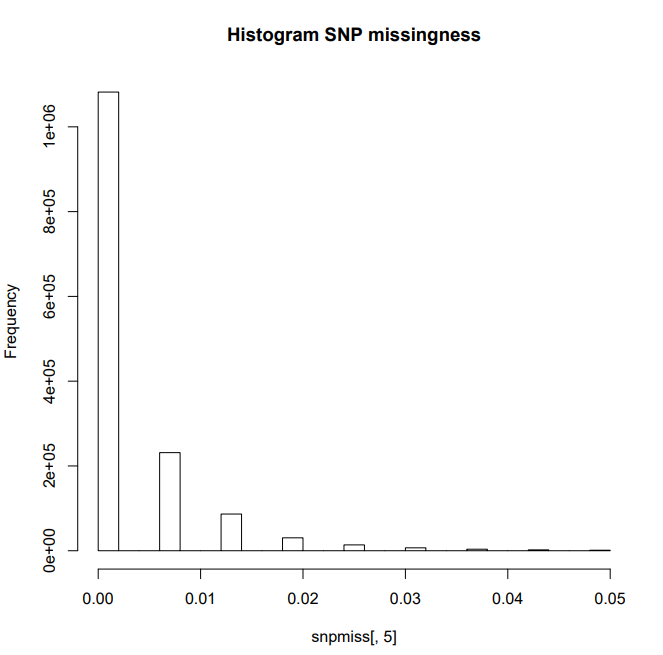
删除高度缺失的 SNP 和个体,对此和所有后续步骤的解释可以在此脚本评论中提到的文章的框 1 和表 1 中找到。
以下两个 QC 命令不会删除任何 SNP 或个体。 但是,良好的做法是使用这些非严格阈值开始 QC。
删除缺失>0.2的SNP。
plink --bfile HapMap_3_r3_1 --geno 0.2 --make-bed --out HapMap_3_r3_2
删除缺失>0.2的个体。
plink --bfile HapMap_3_r3_2 --mind 0.2 --make-bed --out HapMap_3_r3_3
删除缺失>0.02的SNP。
plink --bfile HapMap_3_r3_3 --geno 0.02 --make-bed --out HapMap_3_r3_4
删除缺失>0.02的个体。
plink --bfile HapMap_3_r3_4 --mind 0.02 --make-bed --out HapMap_3_r3_5
3、检查性别差异。
先验确定为女性的受试者的 F 值必须 <0.2,而先验确定为男性的受试者的 F 值必须 >0.8。该 F 值基于 X 染色体近交(纯合性)估计。
不满足这些要求的受试者会被 PLINK 标记为“PROBLEM”。
plink --bfile HapMap_3_r3_5 --check-sex
生成图表以可视化性别检查结果。
Rscript --no-save gender_check.R
gender_check.R
gender <- read.table("plink.sexcheck", header=T,as.is=T)
pdf("Gender_check.pdf")
hist(gender[,6],main="Gender", xlab="F")
dev.off()
pdf("Men_check.pdf")
male=subset(gender, gender$PEDSEX==1)
hist(male[,6],main="Men",xlab="F")
dev.off()
pdf("Women_check.pdf")
female=subset(gender, gender$PEDSEX==2)
hist(female[,6],main="Women",xlab="F")
dev.off()
这些检查表明有一名女性存在性别差异,F 值为 0.99。 (当使用其他数据集时,通常会发现一些差异)。
以下两个脚本可用于处理具有性别差异的个体。
注意,请使用以下两个选项之一来生成 bfile hapmap_r23a_6,我们将在本教程的下一步中使用该文件。
1) 删除有性别差异的个人。
此命令生成状态为"PROBLEM"的个人列表。
grep "PROBLEM" plink.sexcheck| awk '{print$1,$2}'> sex_discrepancy.txt
此命令根据列表删除状态为"PROBLEM"的个人。
plink --bfile HapMap_3_r3_5 --remove sex_discrepancy.txt --make-bed --out HapMap_3_r3_6
2) 估算性别。
plink --bfile HapMap_3_r3_5 --impute-sex --make-bed --out HapMap_3_r3_6
这会将基于基因型信息的性别推算到您的数据集中。
4、生成仅包含常染色体 SNP 的 bfile,并删除具有低次要等位基因频率 (MAF) 的 SNP。
仅选择常染色体 SNP(即从 1 到 22 号染色体)。
awk '{ if ($1 >= 1 && $1 <= 22) print $2 }' HapMap_3_r3_6.bim > snp_1_22.txt
plink --bfile HapMap_3_r3_6 --extract snp_1_22.txt --make-bed --out HapMap_3_r3_7
生成 MAF 分布图。
plink --bfile HapMap_3_r3_7 --freq --out MAF_check
Rscript --no-save MAF_check.R
MAF_check.R
maf_freq <- read.table("MAF_check.frq", header =TRUE, as.is=T)
pdf("MAF_distribution.pdf")
hist(maf_freq[,5],main = "MAF distribution", xlab = "MAF")
dev.off()
去除MAF频率较低的SNP。
plink --bfile HapMap_3_r3_7 --maf 0.05 --make-bed --out HapMap_3_r3_8
1073226 个 SNP 被留下
常规 GWAS 的常规 MAF 阈值介于 0.01 或 0.05 之间,具体取决于样本量。
5、删除不在 Hardy-Weinberg 平衡 (HWE) 中的 SNP。
检查所有 SNP 的 HWE p 值分布。
plink --bfile HapMap_3_r3_8 --hardy
# 选择 HWE p 值低于 0.00001 的 SNP,这是下一个 Rscript 生成的两个图之一所需的,允许放大严重偏离的 SNP。
awk '{ if ($9 <0.00001) print $0 }' plink.hwe>plinkzoomhwe.hwe
Rscript --no-save hwe.R
hwe.R
hwe<-read.table (file="plink.hwe", header=TRUE)
pdf("histhwe.pdf")
hist(hwe[,9],main="Histogram HWE")
dev.off()
hwe_zoom<-read.table (file="plinkzoomhwe.hwe", header=TRUE)
pdf("histhwe_below_theshold.pdf")
hist(hwe_zoom[,9],main="Histogram HWE: strongly deviating SNPs only")
dev.off()
默认情况下,plink 中的 --hwe 选项仅过滤对照组。
因此,我们使用两个步骤,首先我们对对照组使用严格的 HWE 阈值,然后对实验组数据使用不太严格的阈值。
plink --bfile HapMap_3_r3_8 --hwe 1e-6 --make-bed --out HapMap_hwe_filter_step1
案例的 HWE 阈值仅过滤掉与 HWE 极度偏离的 SNP。
这第二个 HWE 步骤只关注实验组,因为在对照组中,所有 HWE p 值 < hwe 1e-6 的 SNP 都已被删除
plink --bfile HapMap_hwe_filter_step1 --hwe 1e-10 --hwe-all --make-bed --out HapMap_3_r3_9
此步骤的理论背景在我们的随附文章中给出:https://www.ncbi.nlm.nih.gov/pubmed/29484742。
6、生成受试者的杂合率分布图并删除杂合率偏离平均值超过 3 sd 的个体。
对一组不高度相关的 SNP 进行杂合性检查。
因此,要生成非(高度)相关的 SNP 列表,我们排除高反转区域(inversion.txt [High LD region])并使用命令 --indep-pairwise 修剪 SNP。
参数[50 5 0.2]分别代表:窗口大小,每一步移动窗口的SNP数量,以及一个SNP同时回归所有其他SNP的多重相关系数。
plink --bfile HapMap_3_r3_9 --exclude inversion.txt --range --indep-pairwise 50 5 0.2 --out indepSNP
运行结束后产生prune.in,prune.out两个文件,prune.in文件中包含的就是通过筛选条件我们需要的SNP位点。文件内容为map文件第二列snp名称(唯一标识符)。
注意,不要删除文件 indepSNP.prune.in,我们将在教程后面的步骤中使用这个文件。
plink --bfile HapMap_3_r3_9 --extract indepSNP.prune.in --het --out R_check
此文件包含您的修剪数据集。
杂合率分布图
Rscript --no-save check_heterozygosity_rate.R
check_heterozygosity_rate.R
het <- read.table("R_check.het", head=TRUE)
pdf("heterozygosity.pdf")
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
hist(het$HET_RATE, xlab="Heterozygosity Rate", ylab="Frequency", main= "Heterozygosity Rate")
dev.off()
以下代码生成与杂合率均值偏差超过 3 个标准差的个体列表。
对于数据操作,我们建议使用 UNIX。但是,在执行统计计算时,R 可能更方便,因此在此步骤中使用 Rscript:
Rscript --no-save heterozygosity_outliers_list.R
heterozygosity_outliers_list.R
het <- read.table("R_check.het", head=TRUE)
het$HET_RATE = (het$"N.NM." - het$"O.HOM.")/het$"N.NM."
het_fail = subset(het, (het$HET_RATE < mean(het$HET_RATE)-3*sd(het$HET_RATE)) | (het$HET_RATE > mean(het$HET_RATE)+3*sd(het$HET_RATE)));
het_fail$HET_DST = (het_fail$HET_RATE-mean(het$HET_RATE))/sd(het$HET_RATE);
write.table(het_fail, "fail-het-qc.txt", row.names=FALSE)
上述命令的输出:fail-het-qc.txt 。
当使用我们的示例数据/HapMap 数据时,此列表包含 2 个个体(即,两个个体的杂合率偏离平均值超过 3 个 SD)。
修改此文件以使其与 PLINK 兼容,方法是删除文件中的所有引号并仅选择前两列。
sed 's/"// g' fail-het-qc.txt | awk '{print$1, $2}'> het_fail_ind.txt
去除杂合率异常值。
plink --bfile HapMap_3_r3_9 --remove het_fail_ind.txt --make-bed --out HapMap_3_r3_10
7、检查你分析的数据集是否有神秘的相关性是很重要的。
假设一个随机人口样本,我们将在本教程中排除所有高于 pihat 阈值 0.2 的个体。
检查 pihat > 0.2 的个体之间的关系。
plink --bfile HapMap_3_r3_10 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2
HapMap 数据集已知包含父子关系。
以下命令将使用 z 值具体可视化这些父子关系。
awk '{ if ($8 >0.9) print $0 }' pihat_min0.2.genome>zoom_pihat.genome
生成一个图来评估关系的类型。
Rscript --no-save Relatedness.R
生成的图在 Hapmap 数据中显示了相当数量的相关个体(扩展图;PO = 父母-后代,UN = 无关个体),这是预期的,因为数据集是这样构建的。
通常,基于家庭的数据应使用特定的基于家庭的方法进行分析。在本教程中,出于演示目的,我们将相关性视为随机人口样本中的隐秘相关性。
在本教程中,我们的目标是从数据集中删除所有“相关性”。
为了证明大部分相关性是由于父母-后代,我们只包括创始人(数据集中没有父母的个人)。
plink --bfile HapMap_3_r3_10 --filter-founders --make-bed --out HapMap_3_r3_11
现在我们将再次寻找 pihat >0.2 的个体。
plink --bfile HapMap_3_r3_11 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2_in_founders
文件 'pihat_min0.2_in_founders.genome' 显示,在排除所有非创始人后,只有 1 个 pihat 大于 0.2 的个体对保留在 HapMap 数据中。
这可能是基于 Z 值的完整同胞或 DZ 双胞胎对。值得注意的是,他们在 HapMap 数据中没有获得相同的家庭身份 (FID)。
对于每对 pihat > 0.2 的“相关”个体,我们建议删除调用率最低的个体。
plink --bfile HapMap_3_r3_11 --missing
使用 UNIX 文本编辑器(例如 vi(m) )检查“相关对”中哪个人的调用率最高。
生成 Pihat 高于 0.2 的个人的 FID 和 IID 列表,以检查谁的通话率较低。
在我们的数据集中,单个 13291 NA07045 的调用率较低。
vi 0.2_low_call_rate_pihat.txt
i
13291 NA07045
按键盘上的 esc!
:X
在键盘上按回车
如果有多个“相关”对,上面生成的列表可以使用与我们单独的“相关”对相同的方法进行扩展。
删除 pihat > 0.2 的“相关”对中调用率最低的个体
plink --bfile HapMap_3_r3_11 --remove 0.2_low_call_rate_pihat.txt --make-bed --out HapMap_3_r3_12
################################################# #################################################
恭喜!! 您刚刚成功完成了第一个教程! 您现在可以进行适当的基因 QC。
对于下一个教程,使用脚本:2_Main_script_MDS.txt,您需要以下文件:
- bfile HapMap_3_r3_12(即,HapMap_3_r3_12.fam、HapMap_3_r3_12.bed 和 HapMap_3_r3_12.bim
质量控制步骤
| 步骤 | 命令 | 功能 | 阈值和解释 |
|---|---|---|---|
| 1. SNP缺失及个体缺失 | --geno | 排除了大部分受试者中缺失的SNP。在此步骤中,低基因型的SNP被删除。 | 建议首先以宽松的阈值(0.2; > 20%)过滤SNP和个体,从而过滤掉缺失程度很高的SNP和个体。再使用更严格的阈值过滤(0.02)。 |
| --mind | 排除基因型缺失率高的个体。在此步骤中,低基因型的个体被移除。 | 注意:在个体移除前应先进行SNP过滤。 | |
| 2. 性别差异 | --check-sex | 根据X染色体杂合/纯合率检查数据集中记录的个体性别与他们自身性别的差异。 | 可以指示样品混合。如果许多研究对象都存在这种差异,则应仔细检查数据。男性的X染色体纯合度估计值为0.8,女性为0.2。 |
| 3. 最小等位基因频率(MAF) | --maf | 仅包括超过MAF阈值的SNP。 | 低MAF的SNP很少见,因此缺乏检测SNP表型关联的能力。这些SNP也更容易出现基因分型错误。MAF阈值应取决于您的样本大小,较大的样本可以使用较低的MAF阈值。对于大样本(N = 100.000)与中等样本(N=10000),通常采用0.01和0.05作为MAF阈值。 |
| 4. 哈迪-温伯格平衡(HWE) | --hwe | 排除偏离哈代-温伯格平衡的指标。 | 基因分型错误的常见指标也可能表明进化选择。 对于二元性状,建议排除:病例中HWE p值<1e-10以及对照组中<1e-6。不大严格的病例阈值可避免丢弃在选择下与疾病相关的SNP。对于数量性状,推荐HWE p值<1e-6。 |
| 5. 杂合性 | 排除杂合率高或低的个体 | 偏差可能表明样品受到污染,近亲繁殖。 建议删除样本杂合率均值偏离±3标准差的个体。 | |
| 6. 相关性 | --genome | 通过下降(IBD)计算所有样本对的同一性。 | 使用独立的SNP(剪枝)进行分析,并将其限制为仅常染色体。 |
| --min | 设置阈值,并创建一个关联性高于所选阈值以上的个体列表。这意味着可以检测到如pihat>0.2的有亲属关系的受试者(即二度亲属)。 | 隐性关联会干扰关联分析。如果你有一个以家庭为基础的样本(例如:父母-后代),则不需要删除相关对,但统计分析应考虑到家庭的相关性。然而,对于基于人群的样本,建议使用pihat阈值为0.2。 | |
| 7. 人口分层 | --genome | 通过下降(IBD)计算所有样品对的同一性。 | 使用独立的SNP(剪枝)进行分析,并将其限制为仅常染色体。 |
| --cluster –mds-plot k | 基于IBS生成数据中任何子结构的k维表示。 | k是需要定义的维数(通常为10)。这是QC的一个重要步骤,它包含多个过程,但为完整起见,此步骤将在“控制人口分层”一节中更详细地描述。 |

















 7251
7251

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









