







拟时序/轨迹分析的基础知识和Monocle2流程可见推文: https://mp.weixin.qq.com/s/aVUpRIkDi83B8_Y_BSBkVA
Monocle3的主要更新:

-
更完善的开发流程:Monocle 3 提供了更好结构化的工作流程,用于学习发育轨迹。
-
支持 UMAP 算法:UMAP 算法用于初始化轨迹推断,为轨迹推断提供了更优的起点。
-
支持多起点的轨迹:能够处理包含多个起点的轨迹推断,从而更好地反映复杂的发育过程。
-
学习含有环路或汇聚点的轨迹:可以学习到包含环形结构或收敛点的复杂轨迹。
-
自动分区算法:使用“近似图抽象”的概念,自动将细胞分区,以学习不相交或并行的轨迹。
-
新的统计测试:为那些具有轨迹相关表达的基因设计了新的统计检验方法,替代旧版的 differentialGeneTest() 和 BEAM()。
-
查询数据集映射到参考集:能够将查询的数据集投射到一个已知的参考集上。
-
注释转移:可以将参考集的注释转移到查询数据集中,便于不同数据集之间的比较和分析。
-
保存和加载 Monocle 对象和转换模型:支持 Monocle 对象和转换模型的保存和加载,方便后续分析。
-
将计数矩阵存储于磁盘:支持将数据存储在磁盘上而不是系统内存中,以节省内存空间。
-
混合负二项分布模型:改进了模型拟合,使用了混合负二项分布,适应更复杂的数据特征。
-
3D 界面:新增 3D 界面,可以用于轨迹和基因表达的可视化,使得数据分析更加直观。

步骤流程
1.导入
示例数据跟monocle2中的一样
rm(list = ls())
library(Seurat)
library(monocle3)
library(dplyr)
load("scRNA.Rdata")
table(Idents(scRNA))
table(scRNA$orig.ident)
head(scRNA@meta.data)
DimPlot(scRNA,label = T)+NoLegend()
2.数据预处理
scRNA$celltype = Idents(scRNA)
levels(Idents(scRNA)) #打出来细胞类型供复制
scRNA = subset(scRNA,idents = c("CD8+ T-cells","NK cells"))
table(Idents(scRNA))
set.seed(1234)
scRNA = subset(scRNA,downsample = 100)
3.提取数据
expression_matrix <- GetAssayData(scRNA, assay = 'RNA',slot = 'counts')
cell_metadata <- scRNA@meta.data
gene_annotation <- data.frame(gene_short_name = rownames(data))
rownames(gene_annotation) <- rownames(data)
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
# 官方文档推荐用稀疏矩阵,如下所示
# cds <- new_cell_data_set(as(umi_matrix, "sparseMatrix"),
# cell_metadata = cell_metadata,
# gene_metadata = gene_metadata)
4.数据处理
# 归一化/预处理数据
# 这一步使用的PCA分析,num_dim数代表纳入的PCA数量
cds <- preprocess_cds(cds, num_dim = 100)
# 这个函数用于确认设定的num_dim数是否足够代表主要变异
plot_pc_variance_explained(cds)
# 去批次,这一步是可选的
# cds <- align_cds(cds, alignment_group = "batch")
# umap降维
cds <- reduce_dimension(cds, preprocess_method = "PCA") # PCA或者LSI
5.plot_cells展示
# color_cells_by 设置展示分组情况
plot_cells(cds,color_cells_by = "celltype")
# genes参数可视化基因的变化
plot_cells(cds, genes=c("TP53","CDK4", "CDK6"))
这张图可以用于确认设定的num_dim数是否代表足够的变异,可以看到在PC大概在15左右就足够了~ 这样子就可以重新对num_dim数进行设置,减少计算资源浪费。

color_cells_by参数设定为celltype之后的细胞展示

genes展示,颜色深浅表示特定基因在该细胞中的表达水平

其中研究者给出了使用UAMP的理由,“快”,当然也可以使用t-SNE。

甚至对于更多的细胞来说,还可以加速分析!需要设置umap.fast_sgd=true

管理批次问题
# 开发者建议在colData中增加批次信息
plot_cells(cds, color_cells_by="orig.ident", label_cell_groups=FALSE)
cds <- align_cds(cds, num_dim = 100, alignment_group = "orig.ident")
cds <- reduce_dimension(cds)
plot_cells(cds, color_cells_by="orig.ident", label_cell_groups=FALSE)
开发者认为批次问题需要严格控制,因此如果下图出现单一样本占多数时,就需要进行去批次处理。

6.将细胞进行聚类分组
cds <- cluster_cells(cds, resolution=1e-5)
plot_cells(cds)
plot_cells(cds, color_cells_by="partition",
group_cells_by="partition")

这个分群的目的是进一步把纳入分析的细胞进行划分(采用community detection方式)。

# cluster_cells之后再安装celltype进行映射一下
plot_cells(cds, color_cells_by="celltype",
label_groups_by_cluster=F)
plot_cells(cds, color_cells_by="celltype",
label_groups_by_cluster=T)
label_groups_by_cluster=F, 之后不会把细胞类型映射到聚类分群之后的细胞上

label_groups_by_cluster=T, 之后会把细胞类型映射到聚类分群之后的细胞上

这个分析在细胞数量和种类多的时候就会有很大的视觉效果。
7.找出每个簇表达的标记基因
marker_test_res <- top_markers(cds, group_cells_by="partition",
reference_cells=1000, cores=8)
top_specific_markers <- marker_test_res %>%
filter(fraction_expressing >= 0.10) %>%
group_by(cell_group) %>%
top_n(3, pseudo_R2) #pseudo_R2是一种排序方法
top_specific_marker_ids <- unique(top_specific_markers %>%
pull(gene_id))
plot_genes_by_group(cds,
top_specific_marker_ids,
group_cells_by="partition",
ordering_type="maximal_on_diag",
max.size=3)
跟上面的图进行对比,聚类一共分了四群,其中1/4是成纤维细胞,2/3是B细胞。然后选择了每一个亚群中表达排序前3的基因进行展示。

8. 对亚群重新命名
colData(cds)$assigned_cell_type <- as.character(partitions(cds))
colData(cds)$assigned_cell_type <- dplyr::recode(colData(cds)$assigned_cell_type,
"1"="F1",
"2"="B1",
"3"="B2",
"4"="F4"
)
plot_cells(cds, group_cells_by="partition", color_cells_by="assigned_cell_type")

9. 若有细胞群分的不明显
cds_subset <- choose_cells(cds)
自动跳转选择起点的页面,可以把混杂细胞框选出来进一步分析(本次分析内容没有明显混杂,使用的图片是官方示例文件)

# 探索混杂细胞群中的差异基因
pr_graph_test_res <- graph_test(cds_subset, neighbor_graph="knn", cores=8)
pr_deg_ids <- row.names(subset(pr_graph_test_res, morans_I > 0.01 & q_value < 0.05))
# 将具有相似表达模式的基因分组到模块
gene_module_df <- find_gene_modules(cds_subset[pr_deg_ids,], resolution=1e-3)
# 绘制这些模块的聚集表达值/揭示不同细胞表达的不同模式
plot_cells(cds_subset, genes=gene_module_df,
show_trajectory_graph=FALSE,
label_cell_groups=FALSE)

想要进一步区分混杂细胞可以探索每个模块中的基因并对它们进行富集分析
这部分的参数来自官网数据,需要自行修改!!
官方文件中也推荐采用他们开发的自动注释的方式——Garnett
cds_subset <- cluster_cells(cds_subset, resolution=1e-2)
plot_cells(cds_subset, color_cells_by="cluster")
colData(cds_subset)$assigned_cell_type <- as.character(clusters(cds_subset)[colnames(cds_subset)])
colData(cds_subset)$assigned_cell_type <- dplyr::recode(colData(cds_subset)$assigned_cell_type,
"1"="Sex myoblasts",
"2"="Somatic gonad precursors",
"3"="Vulval precursors",
"4"="Sex myoblasts",
"5"="Vulval precursors",
"6"="Somatic gonad precursors",
"7"="Sex myoblasts",
"8"="Sex myoblasts",
"9"="Ciliated neurons",
"10"="Vulval precursors",
"11"="Somatic gonad precursor",
"12"="Distal tip cells",
"13"="Somatic gonad precursor",
"14"="Sex myoblasts",
"15"="Vulval precursors")
plot_cells(cds_subset, group_cells_by="cluster", color_cells_by="assigned_cell_type")
10.轨迹图
cds <- cluster_cells(cds, resolution=1e-5)
cds <- learn_graph(cds)
plot_cells(cds, color_cells_by = "partition", # color_cells_by可以修改
label_groups_by_cluster=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE)
# 降低维度并可视化
# 这里其实就是产生轨迹图之后+基因,跟上面的plot_cells很相似
cds <- reduce_dimension(cds)
plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = "celltype")
ciliated_genes <- c("TP53","CDK4", "CDK6")
plot_cells(cds,
genes=ciliated_genes,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)
# 同样还有聚类
cds <- cluster_cells(cds)
plot_cells(cds, color_cells_by = "partition")

# 用order_cells进行起点排序
cds <- cluster_cells(cds, resolution=1e-5)
cds <- learn_graph(cds)
cds <- order_cells(cds)
plot_cells(cds,
color_cells_by = "celltype",
label_cell_groups=FALSE,
label_leaves=TRUE,
label_branch_points=TRUE,
graph_label_size=1.5)
起点可以自定,要结合先验知识!!! 其实会变得很主观。

pseudotime
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5)

自动选择轨迹起点
这个方法适用于自己数据集中包含时间信息(比如有时间分组)
# 假设我的数据集中seurat_clusters是时间信息,请注意是假设!!!
head(colData(cds))
# DataFrame with 6 rows and 11 columns
# orig.ident nCount_RNA nFeature_RNA percent.mt percent.rp percent.hb RNA_snn_res.0.5 seurat_clusters celltype Size_Factor
# <character> <numeric> <integer> <numeric> <numeric> <numeric> <factor> <factor> <factor> <numeric>
# AAAGGTACAATTGGTC-1_1 sample1 3806 1215 7.777194 37.1256 0.00000000 2 2 B-cells 0.645389
# AAAGTGATCTCAACCC-1_1 sample1 3111 1328 6.010929 28.9939 0.06428801 2 2 B-cells 0.527537
# AACAACCTCGTTTACT-1_1 sample1 12631 3134 4.409785 26.5062 0.00791703 8 8 B-cells 2.141857
# AACAAGACATAATCCG-1_1 sample1 5366 1616 4.789415 28.5315 0.00000000 2 2 B-cells 0.909920
# AAGCCATTCTAGACCA-1_1 sample1 22292 4632 0.103176 18.8453 0.00000000 1 1 Fibroblasts 3.780086
# AAGCGAGCAGGTCCGT-1_1 sample1 5689 464 0.123044 4.5878 0.00000000 8 8 B-cells 0.964692
# assigned_cell_type
# <character>
# AAAGGTACAATTGGTC-1_1 B1
# AAAGTGATCTCAACCC-1_1 B1
# AACAACCTCGTTTACT-1_1 B1
# AACAAGACATAATCCG-1_1 B1
# AAGCCATTCTAGACCA-1_1 F1
# AAGCGAGCAGGTCCGT-1_1 B2
# 以下是官网给的代码
# time_bin 官网设定是130-170
get_earliest_principal_node <- function(cds, time_bin="2"){
cell_ids <- which(colData(cds)[, "seurat_clusters"] == time_bin)
closest_vertex <-cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <- igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
cds <- order_cells(cds,
root_pr_nodes=get_earliest_principal_node(cds))
plot_cells(cds, color_cells_by = "pseudotime",
label_cell_groups=FALSE,label_leaves=FALSE,
label_branch_points=FALSE,graph_label_size=1.5)

time_bin的含义:这个参数用于指定分析中的特定时间窗口。在单细胞实验中,尤其是那些涉及时间序列的实验,研究者可能会根据细胞的采样时间将细胞分组到不同的时间窗口(bins)。time_bin 参数就是用来表示这些时间窗口的标签。例如,"130-170" 可能指的是某个具体的发育阶段或实验条件下的时间区间。
所以time_bin是需要咱们的先验信息的! 也就是说要是没这个信息,也没法自动计算起始点。笔者的数据没有时间信息,用了seurat_clusters假装时间信息!

3D轨迹
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d)
cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))
cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by="celltype")
cds_3d_plot_obj
官网图片很好看的,笔者的数据比较丑。

11.基因差异表达分析
一共两种方法:回归分析/用于比较聚类的图自相关分析
其中回归分析需要有时间信息!
用于比较聚类的图自相关分析(Graph-autocorrelation analysis for comparing clusters)不需要时间
# 提取含有B的细胞
cds_test <- cds[,grepl("B", colData(cds)$assigned_cell_type, ignore.case=TRUE)]
# check图像
plot_cells(cds_test, color_cells_by="partition")
# graph_test(自空间自相关分析的统计数据)
pr_graph_test_res <- graph_test(cds_test, neighbor_graph="knn", cores=8)
pr_deg_ids <- row.names(subset(pr_graph_test_res, q_value < 0.05))
# 寻找共调控基因的模块
gene_module_df <- find_gene_modules(cds_test[pr_deg_ids,], resolution=1e-2)
cell_group_df <- tibble::tibble(cell=row.names(colData(cds_test)),
cell_group=partitions(cds)[colnames(cds_test)])
agg_mat <- aggregate_gene_expression(cds_test, gene_module_df, cell_group_df)
row.names(agg_mat) <- stringr::str_c("Module ", row.names(agg_mat))
colnames(agg_mat) <- stringr::str_c("Partition ", colnames(agg_mat))
pheatmap::pheatmap(agg_mat, cluster_rows=TRUE, cluster_cols=TRUE,
scale="column", clustering_method="ward.D2",
fontsize=6)
pr_deg_ids 得到是一群差异基因!!然后把这个基因进行模块化分析,之后提取差异模块并映射在不同簇中进行可视化
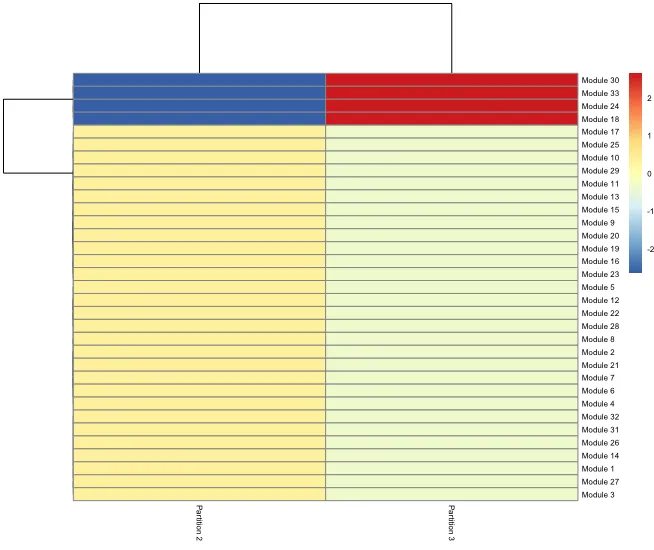
# 查看module
plot_cells(cds_test,
genes=gene_module_df %>% filter(module %in% c(18, 24, 30, 33)),
group_cells_by="partition",
color_cells_by="partition",
show_trajectory_graph=FALSE)
要注意这个图上顶部的数字和图中的数字是不同概念,图片顶部的数字代表module情况(gene_module_df),图中的数字代表不同细胞分组(cell_group_df)。

寻找随伪时间而变化的基因
# 处理数据
# cds <- new_cell_data_set(expression_matrix,
# cell_metadata = cell_metadata,
# gene_metadata = gene_annotation)
# cds <- preprocess_cds(cds, num_dim = 50)
# cds <- align_cds(cds)
# cds <- reduce_dimension(cds)
# cds <- cluster_cells(cds)
# cds <- learn_graph(cds)
# cds <- order_cells(cds)
# plot_cells(cds,
# color_cells_by = "cell.type",
# label_groups_by_cluster=FALSE,
# label_leaves=FALSE,
# label_branch_points=FALSE)
ciliated_cds_pr_test_res <- graph_test(cds, neighbor_graph="principal_graph", cores=8)
pr_deg_ids <- ciliated_cds_pr_test_res %>%
filter(q_value < 0.05) %>%
arrange(-morans_I)
# Moran's I 统计量是衡量一个变量在空间分布上是否表现出系统性变化(即空间相关性)的方法。其值范围通常在 -1(完全离散)到 +1(完全集聚)之间,值为0表示随机分布
plot_cells(cds, genes=c("TP53","CDK4","CDK6"),
show_trajectory_graph=T,
label_cell_groups=T,
label_leaves=T)
# 里边T/F的选择可以自行探索

基因沿单一路径的动态图
test_genes <- c("TP53","CDK4","CDK6")
test_lineage_cds <- cds[rowData(cds)$gene_short_name %in% test_genes,
colData(cds)$celltype %in% c("Fibroblasts")]
test_lineage_cds <- order_cells(test_lineage_cds)
plot_genes_in_pseudotime(test_lineage_cds,
color_cells_by="seurat_clusters",
min_expr=0.5)
图片诡异的原因是笔者的数据里面没有包含时间信息!!!笔者的数据没有时间信息,用了seurat_clusters假装时间信息!
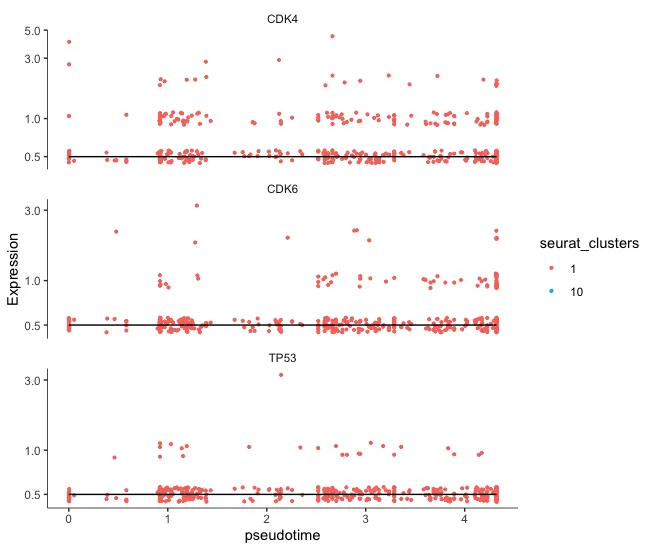
总体感觉这个工具还是不够science的..分析过程中主观性/可人为操作性很强,这是优点也是缺点吧..
如果自己的数据中涉及到时间信息那么这个工具还算是有用处,不然纯粹是一个“绘图工具”。
参考资料
1、monocle3: https://cole-trapnell-lab.github.io/monocle3/docs/introduction/
2、单细胞天地: https://mp.weixin.qq.com/s/siD_cLk4VGD_L-UEIQkm0g
3、Hayley: https://www.jianshu.com/p/c402b6588e17
致谢:感谢曾老师、小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 3766
3766

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









