







10X数据
单个10X样本读取
rm(list = ls())
library(Seurat)
list.files("input/")
# [1] "barcodes.tsv.gz" "features.tsv.gz" "matrix.mtx.gz"
ct = Read10X("input/")
dim(ct)
# [1] 33538 8931
需要把barcodes, features, matrix三个文件放入同一个文件夹中,这三个文件构成一个样本数据。
然后用函数Read10X读取该文件夹。

多个10X样本读取
# 解压缩获取数据
# 一般下载下来的都是tar结尾的压缩文件
untar("GSE185965_RAW.tar",exdir = "GSE185965_RAW")
list.files("GSE185965_RAW/")
# [1] "GSM5627944_HNP210804_barcodes.tsv.gz" "GSM5627944_HNP210804_features.tsv.gz"
# [3] "GSM5627944_HNP210804_matrix.mtx.gz" "GSM5678434_HNP210915_barcodes.tsv.gz"
# [5] "GSM5678434_HNP210915_features.tsv.gz" "GSM5678434_HNP210915_matrix.mtx.gz"
# [7] "GSM5678435_HNP210929_barcodes.tsv.gz" "GSM5678435_HNP210929_features.tsv.gz"
# [9] "GSM5678435_HNP210929_matrix.mtx.gz"
# 可以看到有三个样本(GSM5627944,GSM5678435,GSM5678434)
# 获取每个文件的路径和名称
library(stringr)
fs = paste0("GSE185965_RAW/",dir("GSE185965_RAW/"))
fs
# 这种方式能够把每个文件中的完整读取路径给制作出来
# [1] "GSE185965_RAW/GSM5627944_HNP210804_barcodes.tsv.gz"
# [2] "GSE185965_RAW/GSM5627944_HNP210804_features.tsv.gz"
# [3] "GSE185965_RAW/GSM5627944_HNP210804_matrix.mtx.gz"
# [4] "GSE185965_RAW/GSM5678434_HNP210915_barcodes.tsv.gz"
# [5] "GSE185965_RAW/GSM5678434_HNP210915_features.tsv.gz"
# [6] "GSE185965_RAW/GSM5678434_HNP210915_matrix.mtx.gz"
# [7] "GSE185965_RAW/GSM5678435_HNP210929_barcodes.tsv.gz"
# [8] "GSE185965_RAW/GSM5678435_HNP210929_features.tsv.gz"
# [9] "GSE185965_RAW/GSM5678435_HNP210929_matrix.mtx.gz"
# 把每个文件中的关键词提取出来(要代表每个组别的)
# 获取每个文件的名称,提取关键部分然后去重
samples = dir("GSE185965_RAW/") %>%
str_split_i(pattern = "_",i = 1) %>%
unique()
samples
# [1] "GSM5627944" "GSM5678434" "GSM5678435"
# str_split_i是str_split的拓展用法(stringr包)
# 这个函数处理一个字符向量,每个元素分割成多个部分并提取第 i 个值,返回一个字符向量。这对于从多个类似结构的字符串中快速提取相同位置的元素特别有用。
# 创建存放不同样本的文件夹
ctr = function(s){
# 表示文件路径(创建一个01_data并按照输入的s名称创建文件夹)
ns = paste0("01_data/",s)
if(!file.exists(ns))
dir.create(ns,recursive = T)
}
# 反复运行这个ctr函数
lapply(samples, ctr)
list.files("01_data/")
# 得到三个文件夹
# [1] "GSM5627944" "GSM5678434" "GSM5678435"
# 把每个样本的三个文件复制到单独的文件夹中
lapply(fs, function(s){
for(i in 1:length(samples)){
# 下面的代码是检查一个文件名是否包含特定的样本标识符,如果包含,就将该文件复制到相应的目录中
if(str_detect(s,samples[[i]])){
file.copy(s,paste0("01_data/",samples[[i]]))
}
}
})
list.files("01_data/GSM5627944/") # 展示其中一个
# [1] "GSM5627944_HNP210804_barcodes.tsv.gz" "GSM5627944_HNP210804_features.tsv.gz"
# [3] "GSM5627944_HNP210804_matrix.mtx.gz"
on = paste0("01_data/",dir("01_data/",recursive = T));on
# 获取01_data下边所有文件的名称
# [1] "01_data/GSM5627944/GSM5627944_HNP210804_barcodes.tsv.gz"
# [2] "01_data/GSM5627944/GSM5627944_HNP210804_features.tsv.gz"
# [3] "01_data/GSM5627944/GSM5627944_HNP210804_matrix.mtx.gz"
# [4] "01_data/GSM5678434/GSM5678434_HNP210915_barcodes.tsv.gz"
# [5] "01_data/GSM5678434/GSM5678434_HNP210915_features.tsv.gz"
# [6] "01_data/GSM5678434/GSM5678434_HNP210915_matrix.mtx.gz"
# [7] "01_data/GSM5678435/GSM5678435_HNP210929_barcodes.tsv.gz"
# [8] "01_data/GSM5678435/GSM5678435_HNP210929_features.tsv.gz"
# [9] "01_data/GSM5678435/GSM5678435_HNP210929_matrix.mtx.gz"
nn = str_remove(on, "GSM\\d+_HNP\\d+_");nn
# [1] "01_data/GSM5627944/barcodes.tsv.gz" "01_data/GSM5627944/features.tsv.gz"
# [3] "01_data/GSM5627944/matrix.mtx.gz" "01_data/GSM5678434/barcodes.tsv.gz"
# [5] "01_data/GSM5678434/features.tsv.gz" "01_data/GSM5678434/matrix.mtx.gz"
# [7] "01_data/GSM5678435/barcodes.tsv.gz" "01_data/GSM5678435/features.tsv.gz"
# [9] "01_data/GSM5678435/matrix.mtx.gz"
file.rename(on,nn)
on
# [1] "01_data/GSM5627944/barcodes.tsv.gz" "01_data/GSM5627944/features.tsv.gz"
# [3] "01_data/GSM5627944/matrix.mtx.gz" "01_data/GSM5678434/barcodes.tsv.gz"
# [5] "01_data/GSM5678434/features.tsv.gz" "01_data/GSM5678434/matrix.mtx.gz"
# [7] "01_data/GSM5678435/barcodes.tsv.gz" "01_data/GSM5678435/features.tsv.gz"
# [9] "01_data/GSM5678435/matrix.mtx.gz"
# 修改文件名时,路径时要从工作目录之下开始写的,比如01_data/GSM5627944/GSM5627944_HNP210804_barcodes.tsv.gz可以,只写GSM5627944_HNP210804_barcodes.tsv.gz就不行。
#
# \\d是正则表达式里的任意数字,+代表匹配一次或多次,所以就会把所有的前缀删掉。
# 换不同数据时需要自行改动。正则表达式可以让AI写,然后用Rstudio检验是否达到了目的
# 不要任何犹豫!就写绝对路径导入目标文件下的不同样本文件名
dir='/Users/zaneflying/Desktop/train/01_data/'
samples=list.files(dir)
samples
# [1] "GSM5627944" "GSM5678434" "GSM5678435"
library(data.table)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce=CreateSeuratObject(counts = Read10X(file.path(dir,pro) ) ,
project = pro ,
min.cells = 5,
min.features = 300,)
return(sce)
})
# 把scrList的每个样本进行命名
names(sceList) = samples
# 合并样本
sce.all <- merge(sceList[[1]], y= sceList[ -1 ] ,
add.cell.ids = samples)
dim(sce.all)
# [1] 21703 3273
原数据
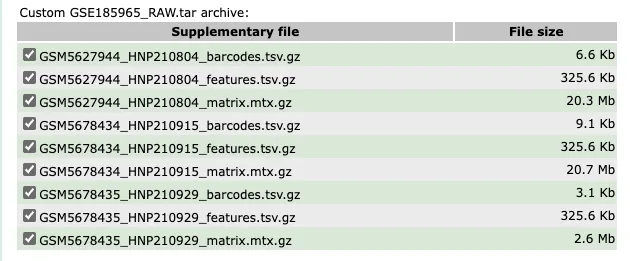
最后结果

h5数据
单个h5数据样本读取
pro = "train"
list.files("input/")
# [1] "GSM8128607_P1_B_filtered_feature_bc_matrix.h5"
sce=CreateSeuratObject(counts = Read10X_h5("input/GSM8128607_P1_B_filtered_feature_bc_matrix.h5") ,
project = pro ,
min.cells = 5,
min.features = 300,)
dim(sce)
#[1] 23062 11442
多个h5数据样本读取
dir='/Users/zaneflying/Desktop/train/GSE260953_RAW/'
samples=list.files( dir )
samples
# [1] "GSM8128607_P1_B_filtered_feature_bc_matrix.h5" "GSM8128608_P1_B2_filtered_feature_bc_matrix.h5"
# [3] "GSM8128609_P1_T_filtered_feature_bc_matrix.h5" "GSM8128610_P1_T2_filtered_feature_bc_matrix.h5"
# [5] "GSM8128611_P2_B_filtered_feature_bc_matrix.h5" "GSM8128612_P2_B2_filtered_feature_bc_matrix.h5"
# [7] "GSM8128613_P2_T_filtered_feature_bc_matrix.h5" "GSM8128614_P2_T2_filtered_feature_bc_matrix.h5"
sceList = lapply(samples,function(pro){
print(pro)
sce =CreateSeuratObject(counts = Read10X_h5( file.path(dir,pro)) ,
project = gsub('_filtered_feature_bc_matrix.h5','',gsub('^GSM[0-9]*_','',pro) ) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
sceList
samples
#整合数据
sce.all=merge(x=sceList[[1]],y=sceList[ -1 ],
add.cell.ids = gsub('_filtered_feature_bc_matrix.h5','',gsub('^GSM[0-9]*_','',samples)))
dim(sce.all)
# [1] 27747 78306
原数据
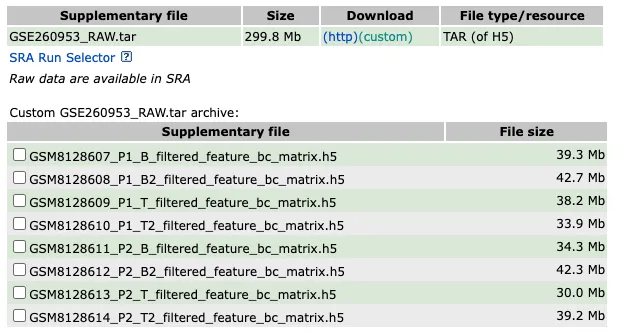
sceList中的project修改的是每个样本的orig.ident
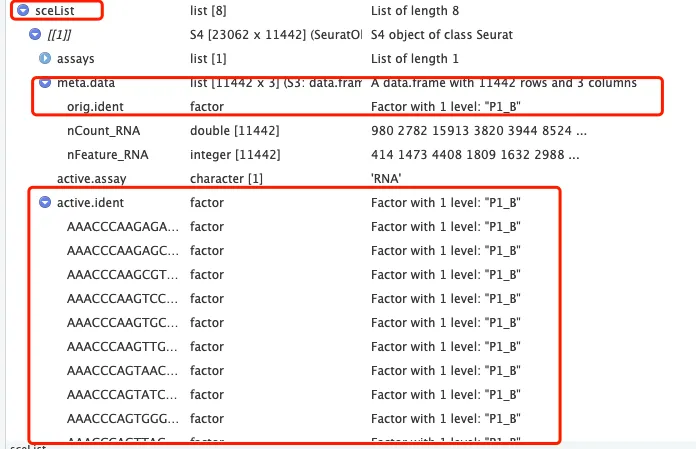
sce.all合并样本之后的add.cell.ids是给每个细胞增加一个标签
txt/csv/tsv数据
单个txt/csv/tsv数据读取
读取数据的函数为fread这个函数很强大,这三种格式的数据都能读取~
所以代码基本不需要修改就可以直接使用~
pro = "train"
list.files("input/")
#[1] "GSE181919_UMI_counts.txt.gz"
ct=fread("input/GSE181919_UMI_counts.txt.gz",data.table = F)
rownames(ct) <- ct$V1
ct <- ct[,-1]
head(ct)[1:5,1:2]
# AAACGGGCATGACGGA.1 AAAGATGAGCAGACTG.1
# RP11-34P13.7 0 0
# FO538757.2 1 3
# AP006222.2 0 0
# RP4-669L17.10 0 0
# RP11-206L10.9 0 0
sce=CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300,)
dim(sce)
多个txt/csv/tsv文件读取
untar("GSE167297_RAW.tar",exdir = "GSE167297_RAW")
dir='/Users/zaneflying/Desktop/train/GSE167297_RAW/'
samples=list.files( dir )
samples
# [1] "GSM5101019_Pt3_Superficial_CountMatrix.txt.gz" "GSM5101020_Pt3_Deep_CountMatrix.txt.gz"
# [3] "GSM5101021_Pt4_Normal_CountMatrix.txt.gz"
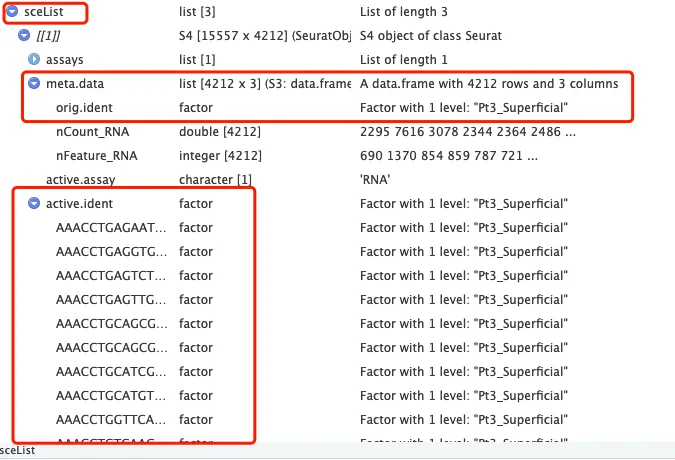
sceList = lapply(samples,function(pro){
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce=CreateSeuratObject(counts = ct ,
project = gsub('_CountMatrix.txt.gz','',gsub('^GSM[0-9]*_','',pro) ) ,
min.cells = 5,
min.features = 300)
return(sce)
})
sceList
samples
#整合数据
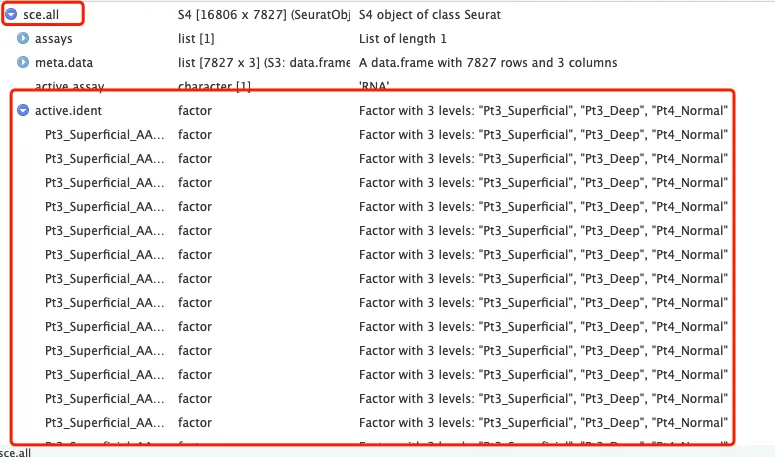
sce.all=merge(x=sceList[[1]],y=sceList[ -1 ],
add.cell.ids = gsub('_CountMatrix.txt.gz','',gsub('^GSM[0-9]*_','',samples)))
dim(sce.all)
# [1] 16806 7827
原数据
sceList中修改proj的意义
sce.all中add.cell.ids的意义
参考资料:
1、生信技能树:
https://mp.weixin.qq.com/s/O-ekjxVqtkv3y-I5HTcoGg
https://mp.weixin.qq.com/s/URu-4l97g18zDmHTrBhBEg
2、单细胞天地: https://mp.weixin.qq.com/s/mpEQU_aKcaq3cbzh8JsSBw
3、生信菜鸟团: https://mp.weixin.qq.com/s/qMi6RAPnhJs-OcqCRotiPw
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 818
818

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









