







重读上一篇关于monocle3的推文的时候感觉内容冗长繁琐,因此笔者把关键部分代码稍作了整理。
推文链接:单细胞拟时序/轨迹分析monocle3流程学习和整理 https://mp.weixin.qq.com/s/NRrFH8sjdUUq20z9hWAFyQ
也可以看一看monocle2推文: 单细胞拟时序/轨迹分析原理及monocle2流程学习和整理 https://mp.weixin.qq.com/s/aVUpRIkDi83B8_Y_BSBkVA
分析步骤
1、导入
rm(list = ls())
library(paletteer)
library(Seurat)
library(monocle3)
library(dplyr)
library(BiocParallel)
library(ggplot2)
register(MulticoreParam(workers = 4, progressbar = TRUE))
load("scRNA.Rdata")
# check一下
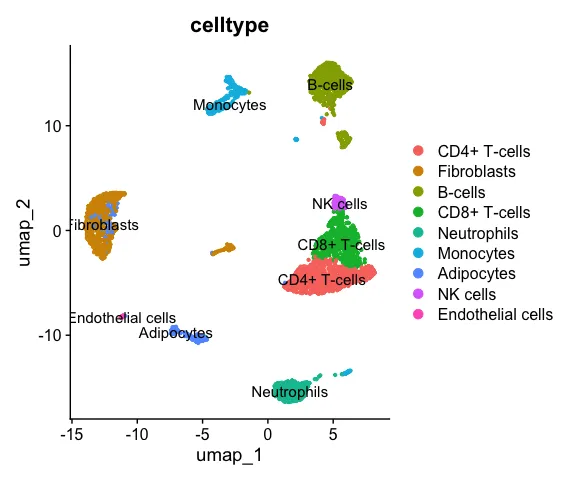
DimPlot(scRNA,pt.size = 0.8,group.by = "celltype",label = T)
2、数据预处理
levels(Idents(scRNA)) #打出来细胞类型供复制
# [1] "CD4+ T-cells" "Fibroblasts" "B-cells"
# [4] "CD8+ T-cells" "Neutrophils" "Monocytes"
# [7] "Adipocytes" "NK cells" "Endothelial cells"
# 重设等级也可以不设
# scRNA$celltype <- factor(scRNA$celltype,
# levels = c("Endothelial cells"))
Idents(scRNA) <- scRNA$celltype
3.提取数据
expression_matrix <- GetAssayData(scRNA, assay = 'RNA',layer = 'counts')
cell_metadata <- scRNA@meta.data
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix))
rownames(gene_annotation) <- rownames(expression_matrix)
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
4.数据处理
# 归一化/预处理数据
cds <- preprocess_cds(cds, num_dim = 100)
# 这个函数用于确认设定的dim数是否足够代表主要变异
plot_pc_variance_explained(cds)
# 降维聚类,可选择UMAP、PCA或者TSNE
cds <- reduce_dimension(cds,reduction_method='UMAP',
preprocess_method = 'PCA')
cds <- cluster_cells(cds) #cluster your cells
5.轨迹推断
# 轨迹推断
cds <- learn_graph(cds,verbose=T,
use_partition=T, #默认是T,T时是顾及全局的情况
)
plot_cells(cds,
color_cells_by = 'celltype',
label_groups_by_cluster=FALSE,
cell_size=1,group_label_size=4,
trajectory_graph_color='#023858',
trajectory_graph_segment_size = 1)

6.定义起点-轨迹可视化
# 定义root cell, 推断拟时方向
# 结合先验知识自定(示例数据)
cds <- order_cells(cds)
# 可视化
plot_cells(cds, label_cell_groups = F,
color_cells_by = "pseudotime",
label_branch_points = F,
graph_label_size = 0,
cell_size=2,
trajectory_graph_color='black',
trajectory_graph_segment_size = 2)
示例数据,起点是随便点的。

7.轨迹差异基因分析
# 提取不同轨迹的差异基因/并选择前12个
# neighbor_graph="principal_graph"提取轨迹上相似位置是否有相关的表达
trace_genes <- graph_test(cds,
neighbor_graph = "principal_graph",
cores = 4)
track_genes_sig <- trace_genes %>%
top_n(n=12, morans_I) %>%
pull(gene_short_name) %>%
as.character()
# 差异基因绘制
levels(Idents(scRNA)) #打出来细胞类型供复制
# [1] "CD4+ T-cells" "Fibroblasts" "B-cells"
# [4] "CD8+ T-cells" "Neutrophils" "Monocytes"
# [7] "Adipocytes" "NK cells" "Endothelial cells"
lineage_cds <- cds[rowData(cds)$gene_short_name %in% track_genes_sig,
colData(cds)$celltype %in% c("NK cells",
"Adipocytes",
"Monocytes")]
#lineage_cds <- order_cells(lineage_cds)
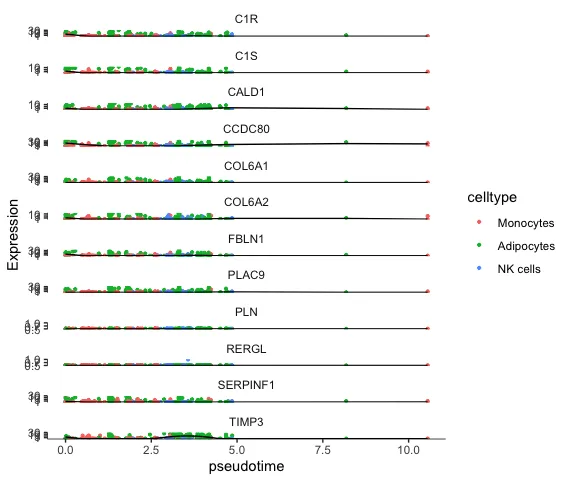
plot_genes_in_pseudotime(lineage_cds,
color_cells_by="celltype",
min_expr=0.5)
# 细胞映射
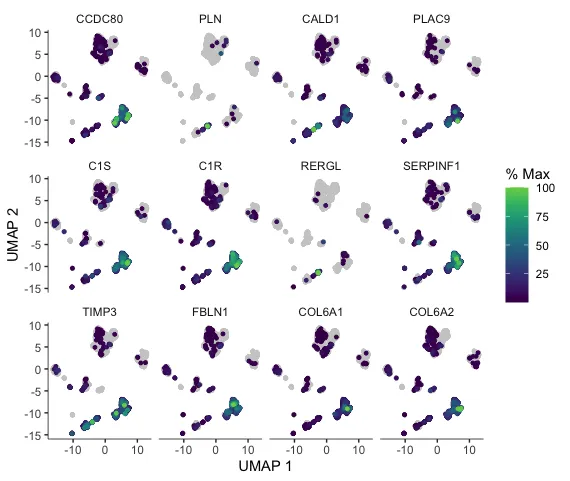
plot_cells(cds, genes= track_genes_sig,
cell_size=1,
show_trajectory_graph=FALSE,
label_cell_groups=FALSE,
label_leaves=FALSE)
morans I 是一种用于测量基因表达的空间自相关性的统计量,判断某个基因的表达是否显示出空间聚集或空间模式。一般会用这个值结合/不结合P值去做基因筛选。得到了基因之后后续就可以做拟时序的热图了
下面的图是不同细胞群中的某一些基因随着时间变化的表达图。其中TIMP3这个基因在2.5-5.0之间出现了一个小的峰,说明在这个时间段的Adipocytes中这个基因表达增加了,但过了这个时间点之后后面表达又降低回去了,并且后面的时间点钟这三种细胞几乎都没有了。
这是不同差异基因的UMAP表达图。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 7158
7158

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









