







 本文详细介绍了如何使用R中的oligo软件包处理Affy和NimbleGen芯片数据,涵盖了从数据下载、软件安装到探针水平分析、表达量计算、P/A过滤、差异表达探针获取以及探针到基因的转换等步骤。通过实例展示了处理过程,并提供了两种探针到基因转换的方法:使用芯片注释文件和基因组特征文件。
本文详细介绍了如何使用R中的oligo软件包处理Affy和NimbleGen芯片数据,涵盖了从数据下载、软件安装到探针水平分析、表达量计算、P/A过滤、差异表达探针获取以及探针到基因的转换等步骤。通过实例展示了处理过程,并提供了两种探针到基因转换的方法:使用芯片注释文件和基因组特征文件。
本博客介绍过 Affy芯片的处理方法 ,其中所使用的软件包有一定的局限性,无法读取和分析一些新版Affy芯片。本文介绍oligo软件包的处理方法以解决这些问题。oligo软件包并不是新出现的软件包,只因新类型芯片的不断推出,关注它的用户越来越多。而且,除了用于Affy芯片处理外,oligo软件包还可处理NimbleGen芯片。
oligo处理芯片的原理和其他方法相同,难点在最后一步:从探针到基因注释的转换。
1 准备工作
1.1 数据下载
本文还是以Affy芯片为例,介绍oligo软件包处理芯片数据的方法。所用数据集为NCBI GSE72154,可通过HTTP或FTP下载原始数据文件。建议使用FTP下载,登录ftp.ncbi.nlm.nih.gov站点,在/geo/datasets下找到GSE72154子目录后全部下载。下载的目录中包含matrix、miniml、soft和suppl四个子目录。
1.2 软件包安装
oligo是bioconductor软件包,可以通过下面语句安装:
source("https://bioconductor.org/biocLite.R") biocLite("oligo")
2 文件读取与信息修改
oligo读取芯片原始数据文件的函数为 read.celfiles 或 read.xysfiles (NimbleGen芯片),可以直接读取压缩文件,一张芯片对应一个文件。使用FTP下载后CEL文件位于suppl子目录下,把GSE72154_RAW.tar解压缩为gz文件或CEL文件即可。
library(oligo) data.dir <- "GSE72154/suppl/" (celfiles <- list.files(data.dir, "\\.gz$"))
## [1] "GSM1856253_Bohmer_685-3_Ler-0_Rep1_AraGene-1-1-st.CEL.gz" ## [2] "GSM1856254_Bohmer_685-4_Ler-0_Rep2_AraGene-1-1-st.CEL.gz" ## [3] "GSM1856255_Bohmer_685-1_wrky43_Rep1_AraGene-1-1-st.CEL.gz" ## [4] "GSM1856256_Bohmer_685-2_wrky43_Rep2_AraGene-1-1-st.CEL.gz"
data.raw <- read.celfiles(filenames = file.path(data.dir, celfiles))
## Reading in : GSE72154/suppl//GSM1856253_Bohmer_685-3_Ler-0_Rep1_AraGene-1-1-st.CEL.gz ## Reading in : GSE72154/suppl//GSM1856254_Bohmer_685-4_Ler-0_Rep2_AraGene-1-1-st.CEL.gz ## Reading in : GSE72154/suppl//GSM1856255_Bohmer_685-1_wrky43_Rep1_AraGene-1-1-st.CEL.gz ## Reading in : GSE72154/suppl//GSM1856256_Bohmer_685-2_wrky43_Rep2_AraGene-1-1-st.CEL.gz
read.celfiles 函数读取数据的过程中会检查系统中是否已安装相应的芯片注释软件包,没有安装的话将有警告。如果在读取的过程中没有自动安装相应注释软件包,请手动安装后再重新读取数据。
本例含4张芯片,文件名上面代码已列出,含野生型和突变体各两个重复。样品名称默认为文件名,需简单修改:
treats <- strsplit("Ler Ler wrky wrky", " ")[[1]] (snames <- paste(treats, 1:2, sep = ""))
## [1] "Ler1" "Ler2" "wrky1" "wrky2"
sampleNames(data.raw) <- snames pData(data.raw)$index <- treats sampleNames(data.raw)
## [1] "Ler1" "Ler2" "wrky1" "wrky2"
pData(data.raw)
## index ## Ler1 Ler ## Ler2 Ler ## wrky1 wrky ## wrky2 wrky
样品名称与芯片文件必需对应,不能有重复;pData的index数据表示处理类型。
3 探针水平分析
原始芯片数据读入后应简单评估一下芯片的质量,这在 其他文章 中介绍过,oligo中也提供了很多方法,如果样品重复量足够,boxplot是比较直观的方法:
fit1 <- fitProbeLevelModel(data.raw) boxplot(fit1, names = NA, col = as.factor(treats)) legend("topright", legend = unique(treats), fill = as.factor(unique(treats)), box.col = NA, bg = "white", inset = 0.01)
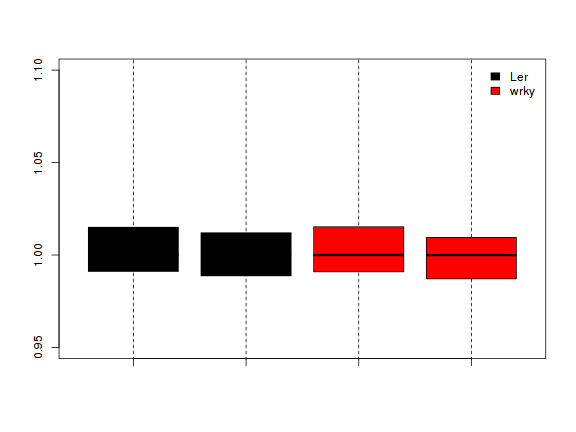
当然也可以使用MAplot:
par(mfrow = c(2, 2)) MAplot(data.raw[, 1:4], pairs = FALSE)
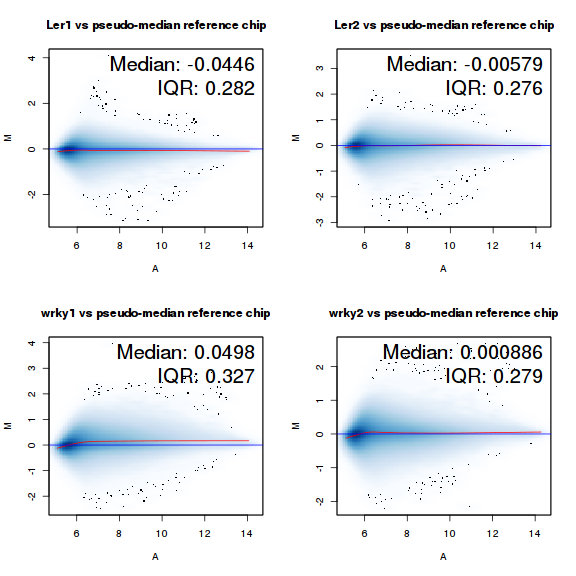
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 2704
2704

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









