







一、软件介绍 DADA2(Divisive Amplicon Denoising Algorithm 2)是一个用于建模和修正多种测序平台(Illumina、Roche 454)测序扩增子错误的开源软件(R)包。在扩增子分析处理流程中,DADA2算法能够准确地推断样本序列并寻找出单个核苷酸的差异(往往能够比其他方法识别出更多真实变体和输出更少的虚假序列)。DADA2是一个通过构建错误率模型来推测扩增子序列是否来自模板的算法,以自身数据的错误模型为参数,不用依赖于其他参数分布模型。DADA2最重要的优势是用了更多的数据,错误模型包含了质量信息,而其他的方法都在过滤低质量之后把序列的质量信息忽略。DADA2的错误模型也包括了定量的丰度,而且该模型也计算了各种不同转置的概率。和比较OTU数据库的聚类算法不同,DADA2采取的是降噪算法。聚类算法与降噪算法的差异与优劣可以参考https:// pubmed.ncbi.nlm.nih.gov/28731476/
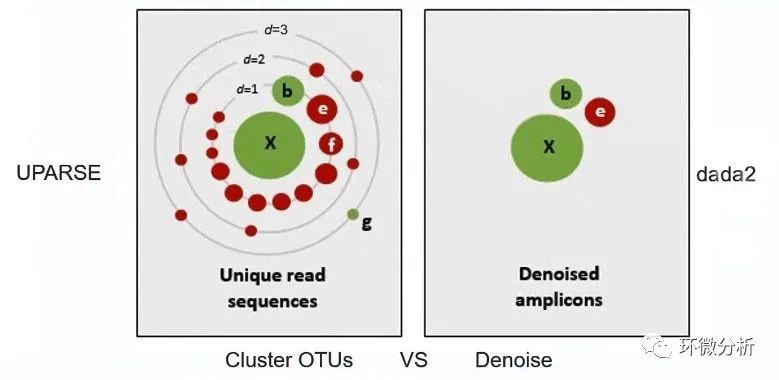
解读:聚类算法通常在97%的相似水平下聚类生成OTU,然后选择每个聚类群(OTU)中最高丰度序列作为代表性序列(如上左图,通过聚类算法获取了众多红色虚假序列与真实序列X、b和g,聚类算法将把众多红色虚假序列与真实序列b、g一并纳入X真实序列中,导致丢失了真实序列b和g);降噪算法是基于丰度与测序原理评估获取程序认为的“真实序列”作为ASV(如上右图,通过聚类算法获取了虚假序列e与真实序列X、b),后期我们可以通过丰度过滤或其他参数设置,尽量识别出e并去除。 二、软件准备 ① 安装R、RStudio和Rtools,打开RStudio
② 获取DADA2包
# 选用国内用户清华镜像站,国内用户加速下载
site="https://mirrors.tuna.tsinghua.edu.cn/CRAN"
# 检查是否存在Biocondoctor安装工具,没有则安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager",repo=site)
# 加载安装工具与安装DADA2,如果提示R包版本与R版本不匹配,可以根据提示信息选择安装合适的DADA2版本
library(BiocManager) BiocManager::install("dada2", version = "3.14")
# 需要显示版本信息
# 加载DADA2包
library(dada2) packageVersion("dada2")
#使用命令“?`dada2-package`” 或”help("dada2-package")”获取DADA2包的帮助文档,针对任何内置或加载获取的R包均可以使用“?具体R包”获取相应函数的帮助文档,包括内置的 命令、参数选择与使用方法。
?help("dada2-package")
三、基本分析流程
官网(DADA2 Pipeline Tutorial (1.16))的DADA2 pipeline Tutorial分析流程详细介绍了如何从原始测序序列分析得到ASV矩阵表格以及物种注释信息,也可获取开源代码,示例数据,R包手册与相关文章。
DADA2处理扩增子测序数据基本流程包括Inspect read quality profiles(检查序列质量文件)、Filter and trim(过滤与裁剪)、Learn the Error Rates(错误率建模)、Sample Inference(样本推断)、Merge paired reads(合并双端序列)、Construct sequence table(构建ASV表格)、Remove chimeras(去除嵌合体)、Track reads through the pipeline(处理流程统计)、Assign taxonomy(注释分类)、Evaluate accuracy(准确性评估)等步骤。
四、分析实操
(1)数据导入
① 数据准备
准备好原数据集.fastq文件和Sliva138注释数据库silva_nr99_v138.1_train_set.fa.gz(Taxonomic reference data)、silva_species_assignment_v138.1.fa.gz(Taxonomic reference data)、SILVA_SSU_r138_2019.RData(http://DECIPHER.codes/Downloads.html),全部存放在目标路径C:\Users\ASUS\Desktop\R\rawdata下,也可以选择其他注释数据库,例如RDP与Greengene,根据自己需求下载即可。
提示:DADA2处理流程执行之前要确保测序数据符合以下三个要求:① 样本已完成拆分,即拆分为单个样本的 fastq 文件;② 已去除非生物核苷酸序列,例如引物、接头、接头等,如果没有,可以使用裁剪命令对序列端口的非生物序列进行定量裁剪;③ 如果是双端测序数据,正向数据和反向数据.fastq 文件应该包含匹配顺序的序列。
② 设置工作路径,即数据所在目录
path <- "C:/Users/ASUS/Desktop/R/rawdata" list.files(path)

③ 获取文件名列表
利用文件的固定命名方式,读取需要的(双端)数据文件名,通过字符串操作函数提取单个样品测序文件信息。其中,list.files返回指定目录中的文件名;pattern指定返回指定类型的文件;full.names = TRUE返回带有路径的完整文件名。
# 返回测序正向文件完整文件名 fnFs <- sort(list.files(path, pattern= "_R1_001.fastq", full.names = TRUE)) # 返回测序反向文件完整文件名 fnRs <- sort(list.files(path, pattern="_R2_001.fastq", full.names = TRUE))

提示:如果文件名格式不同,则需要修改字符串操作。
# 使用命令“?sort()”获取sort()函数的帮助文档,针对任何内置或加载获取的函数均可以使用“?具体函数”获取相应函数的帮助文档
?sort()
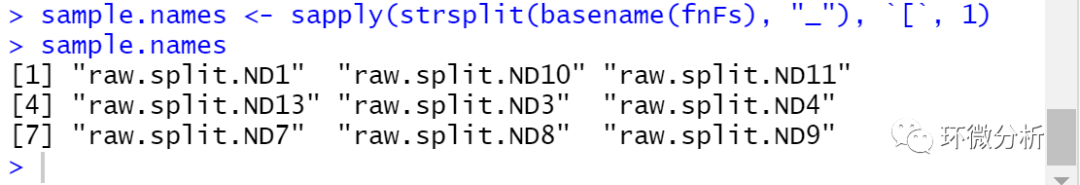
# 提取文件名中第一个`_`分隔的前文本作为样品名 sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1) # 检查提取出的文件名 sample.names
④ 查看序列质量
# 绘制前2个样本的正向序列的碱基质量图 plotQualityProfile(fnFs[1:2])
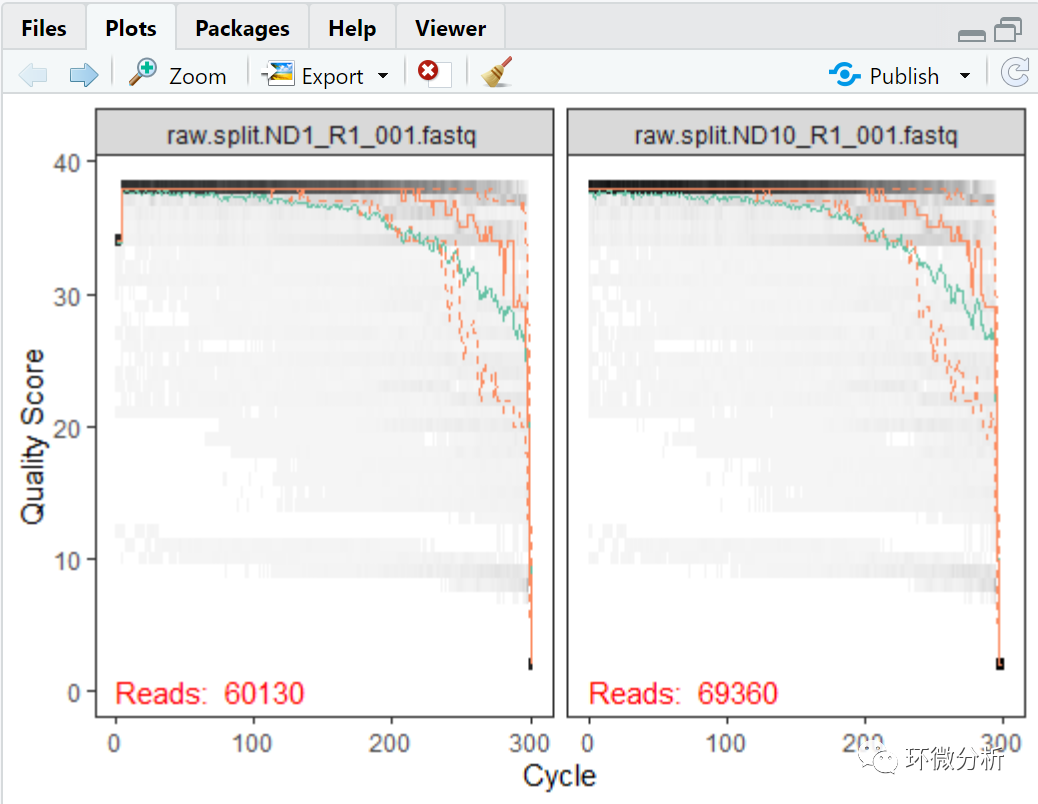
解读:灰度图是每个基本位置上每个质量分数的频率的热图。每个位置的平均质量分数由绿线表示,质量分数分布的四分位数由两条橙色虚线表示。每个位置的质量值中位数由橙色实线表示。如果序列的长度不同,则将绘制一条红线,显示扩展到该位置的序列百分比,这对454焦磷酸测序等技术更有用,因为Illumina读数通常都是相同的长度。正向序列质量前段序列质量较好,后端较差,我们通常建议修剪掉开头的引物与barcode等非生物序列,也会裁剪掉后端错误率比较高的核苷酸序列。反向序列质量往往比正向序列差,特别是在最后,从平均质量值突降的位置进行剪切会提高算法对稀有序列的敏感性。
提示:当我们在处理自己的数据时,不仅仅需要根据质量突降的位置进行裁剪位置的选择,首先我们必须确定测序的双端序列必须拥有足够的重叠(overlap),至少保证裁剪之后在20 bp以上,以保证拼接效率。
(2)序列裁剪与过滤
示例数据为双端测序原始数据,还未去除barcode和引物。去除引物应该在在质量过滤之前(每个碱基都会增加预期误差)和找到唯一序列之前(引物扩增区域的变异会将序列分成数个特异性序列,从而简化特异序列丰度的计算)。已知测序引物为338F(5’-ACTCCTACGGGAGGCAGCAG-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’),引物长度均为20bp,Barcode的长度为6,可以打开数据文件进行验证,以raw.split.N_D_1.1.fq为例:

① 质量过滤裁剪
由输出的碱基质量图可知,序列长度基本在300 bp,引物与barcode加和长度为26 bp,可以通过filterAndTrim命令对正向序列和反向序列分别裁剪掉30和30 bp,maxEE参数表示允许在条reads中期望错误率上限,maxN 是指序列中可以容忍存在不明碱基N的数量,默认没有限制,本次设置为0,truncQ为一条序列第一个碱基允许的最低质量限值,低于这个限值就会被截断,默认为2,;rm.phix默认为TRUE,将去除比对上参考PhiX基因组的序列;compress,默认输出压缩格式的结果;multithread为默认单线程,使用multithread=TRUE提高运算速度。
# 设置过滤文件的输出路径,将过滤后的文件存于\filtered filtFs <- file.path(path, "filtered", paste0(sample.names, "_F_filt.fastq.gz")) filtRs <- file.path(path, "filtered", paste0(sample.names, "_R_filt.fastq.gz")) # 过滤文件输出,统计结果保存于out out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen=c(270,270), maxN=0, maxEE=c(2,2), truncQ=2, rm.phix=TRUE, compress=TRUE, multithread=T) head(out)

② 查看过滤文件
在路径"C:/Users/ASUS/Desktop/R/ rawdata/ filtered "下查看过滤文件
提示1:16S rDNA的结构,从5’端到3’端有10个功能区,即V1-V10。由于V4-V5的特异性较高,所以一般都是测V4区域。如果用的是overlap比较低的数据,比如V1-V2或者V3-V4, truncLen的数值就需要更大。绝大部分参数都可以根据自己需求进行调整,如果我们需要缩短过滤时间,maxEE参数设置更小一些,想要保留多数的reads,那就需要对maxEE参数设置的更大些,尤其是反向测序数据(例如maxEE=c(2,5))。
提示2:对于ITS测序结果,序列长度变化较大,可以考虑不进行裁剪,但是要确保引物在这之前已经被去除干净。
(3)计算错误率
DADA2算法使用机器学习构建参数误差模型(err),认为每个扩增子测序样品都具有不同的误差比率。learnErrors方法通过交替估计错误率和对参考样本序列学习错误模型,直到学习模型同真实错误率收敛于一致。这是DADA2中运行最耗费计算资源的一步,8核16线程笔记本电脑运行约20-30分钟。
# 分别计算正向和反向序列错误率 errF <- learnErrors(filtFs, multithread=TRUE) errR <- learnErrors(filtRs, multithread=TRUE)
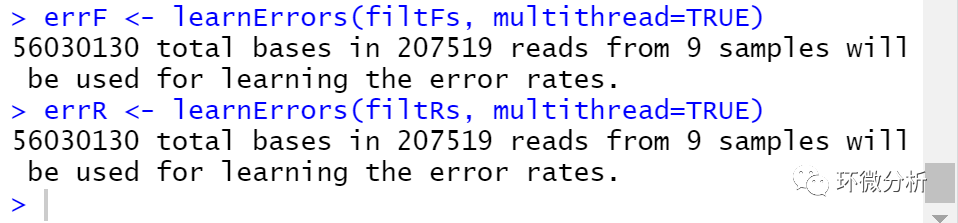
# 画出错误率统计图 plotErrors(errF, nominalQ=TRUE)
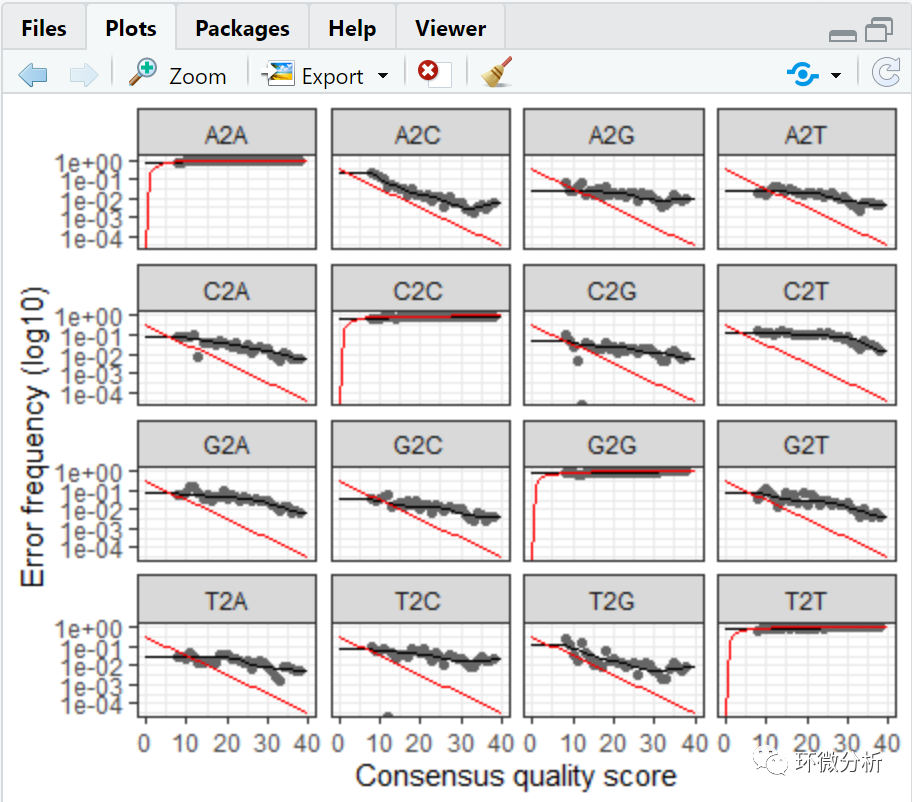
plotErrors(errR, nominalQ=TRUE)
错误率统计图表示了所有可能的错误(A→C,A→G,…),图中点表示观察得到的错误率,黑线表示通过算法学习评估得到的错误率,红色的曲线表示由Q-score的定义下预期的错误率。这里估计的错误率(黑线)同观察到的错误率(点)拟合程度很好,并且错误率随着预期质量下降而下降。
提示:DADA2核心算法亦是参数学习,计算量非常可观。面对如此巨大的数据和需要消耗的计算资源,这一模型的展示便不适合我们实际较大的数据量,可以通过增加nbase参数调整拟合程度以减少计算量。
(4)去冗余(去除重复序列)
去重复是将所有相同的测序读数组合成“独特序列”,其相应的“丰度”等于具有该独特序列的读数的数量。与usearch去冗余步骤类似,仅仅保留重复序列中的一条序列,大量节省计算资源。DADA2保留了去重序列的质量信息,这些质量信息取自重复序列的均值。这一信息文件将作为参考错误模型用于后续序列处理,以提高了DADA2算法准确性。
# 去除正向序列数据中的重复序列 derepFs <- derepFastq(filtFs, verbose=TRUE)
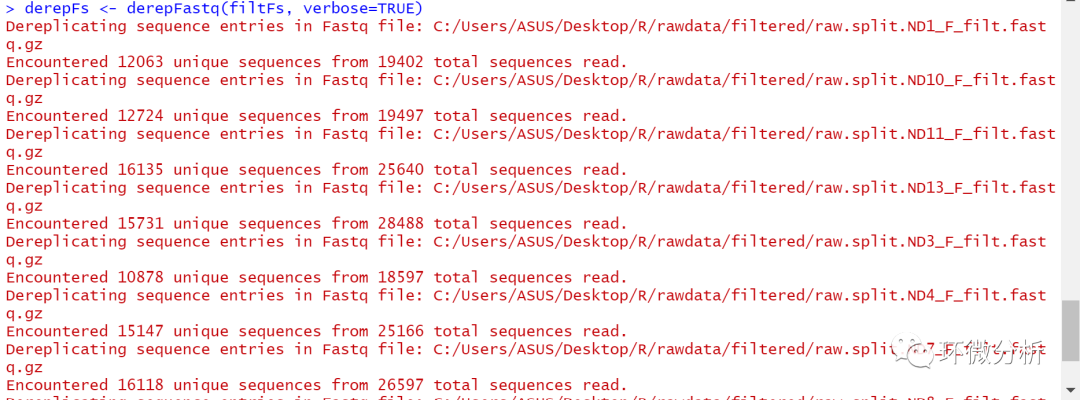
# 去除反向序列数据中的重复序列 derepRs <- derepFastq(filtRs, verbose=TRUE)
提示:如果数据量很大,可能需要使用别的策略更为妥当,参考A DADA2 workflow for Big Data (1.4 or later)。这个命令是一个批量的操作,如果出现处理大文件过程内存不足的情况,可以考虑逐个处理每个样本。
(5)DADA2核心算法(步骤)
DADA2算法是一种分裂式分割算法。首先,将每个reads全部看作单独的单元,sequence相同的reads被纳入一个sequence,reads个数即成为该sequence的丰度(abundance);其次,计算每个sequence丰度的p-value,当最小的p-value低于设定的阈值时,将产生一个新的partition。每一个sequence将会被归入最可能生成该sequence的partition;最后,依次类推,完成分割归并。
# 基于错误模型进一步质控 dadaFs <- dada(derepFs, err=errF, multithread=TRUE) dadaRs <- dada(derepRs, err=errR, multithread=TRUE)
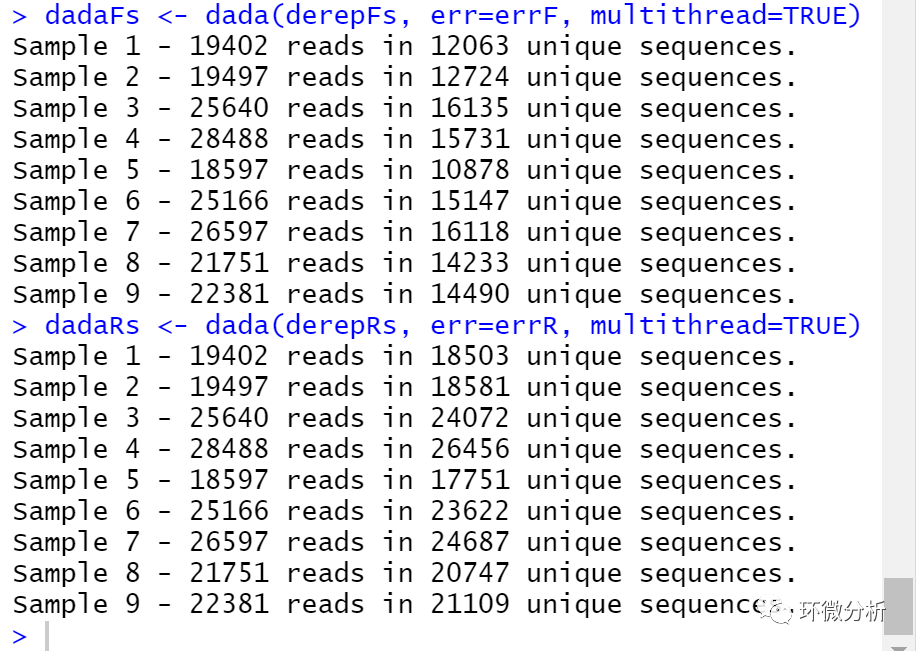
# 查看正向序列dada质控结果
dadaFs[[1]]

以上结果表示第一个正向序列样本通过去冗余获得12063个独特序列,然后通过DADA2算法从12063个独特序列推断出626个真实的物种序列。dada-class返回大量对象,具体参见help(dada-class)参考资料,包括关于每个去噪序列质量的多个评价指标。
提示1:在本示例中,所有样本都同时加载到内存中。如果处理接近或超过可用内存(RAM)的数据集,样品数量较多时,我们需要逐个处理样本。请参阅 DADA2大数据工作流程(A DADA2 workflow for Big Data (1.4 or later))。
提示2:DADA2同时支持454和IonTorrent数据,但是建议对其中一些参数进行修改,可以通过R语言中?setDadaOpt来调取帮助文件,探索这些参数的修改方法。
(6)合并双端序列
我们现在将正向序列和反向序列合并在一起以获得完整的序列。通过将去噪的正向序列与相应的去噪反向序列的反向互补序列比对,然后构建合并的“overlap”进行合并。默认情况下,仅当正向和反向序列重叠至少20个碱基并且在重叠区域中彼此相同时才有效地输出合并序列。 # 合并双端序列 mergers <- mergePairs(dadaFs, derepFs, dadaRs, derepRs, verbose=TRUE)
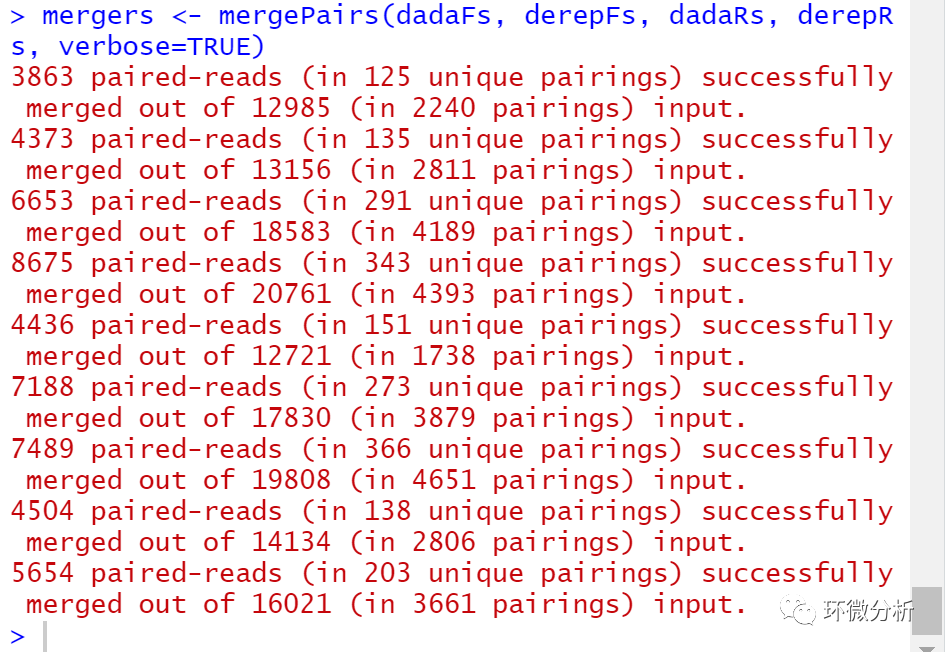
# Inspect the merger data.frame from the first sample
head(mergers[[1]])
提示:mergers对象格式为R语言的数据框,数据框中包含序列及其丰度信息,未能成功合并的序列被删除。没有overlap的数据建议加入参数justConcatenate=TRUE。
(7)构建ASV表
# 构建ASV表,amplicon sequence variant(ASV)表类似于我们传统的OTU表 seqtab <- makeSequenceTable(mergers) # dim()的第二个值为扩增子序列个数,table(nchar())的统计结果表示每个读长下有多少个扩增子序列。 dim(seqtab) # Inspect distribution of sequence lengths 查看序列长度分布 table(nchar(getSequences(seqtab)))

(8)去除嵌合体
Dada核心质控算法去除了大部分错误,但嵌合体仍然存在,去噪后序列的准确性使得识别嵌合体比处理模糊OTU更简单。
#去除嵌合体 seqtab.nochim <- removeBimeraDenovo(seqtab, method="consensus", multithread=TRUE, verbose=TRUE) dim(seqtab.nochim) sum(seqtab.nochim)/sum(seqtab)

嵌合体的数量历来被大家讨论过很多次,因为不同实验,不同样品等等很多的因素都与嵌合体数量有关。进行去除嵌合体步骤后,大量嵌合体被去除了。sum(seqtab.nochim)/sum(seqtab)=0.948表示嵌合体的去除使得剩下全部序列数量的94.8%,如果去除效果不佳,很可能原始数据有问题,大概率是原始数据中模糊核苷酸的引物序列还没被去除。
# 导出生成的ASV表(seqtab.nochim) write.csv(seqtab.nochim,file="C:/Users/ASUS/Desktop/R/rawdata/filtered/ASV.CSV",append = FALSE, quote = FALSE , sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")

(9)物种注释(方法一)
在16S/18S/ITS 扩增子测序中,给序列变体添加物种注释信息是很常见的。DADA2包提供了可以实现本地处理的朴素贝叶斯分类器方法。该assignTaxonomy函数将一组要分类的序列和具有已知分类的参考序列训练集作为输入,并输出通过minBoot检验的注释文件。 # 这里我们的16s序列注释,采用下载Silva参考数据库进行训练和注释,首先从页面 http://benjjneb.github.io/DADA2/training.html 下载相应数据库,有16S可选 Silva 132/128/123,RDP trainset 16/14, Greengene 13.8;真菌ITS选择UNITE;这里我们使用Silva 138,需要下载序列训练器和物种注释参考序列数据包两个文件。
# 用训练器将序列分类,生成的文件将保存至工作目录,如果放在其它位置,请修改下面代码中path变量 taxa <- assignTaxonomy(seqtab.nochim, paste0(path, "/silva_nr99_v138.1_train_set.fa.gz "), multithread=TRUE) # 完成分类后,用参考序列数据包,对应填充数据信息 taxa <- addSpecies(taxa, paste0(path, "/silva_species_assignment_v138.1.fa.gz"))

# 输出taxa文件
write.csv(taxa,file="C:/Users/ASUS/Desktop/R/rawdata/filtered/taxa.CSV",append = FALSE, quote = FALSE , sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
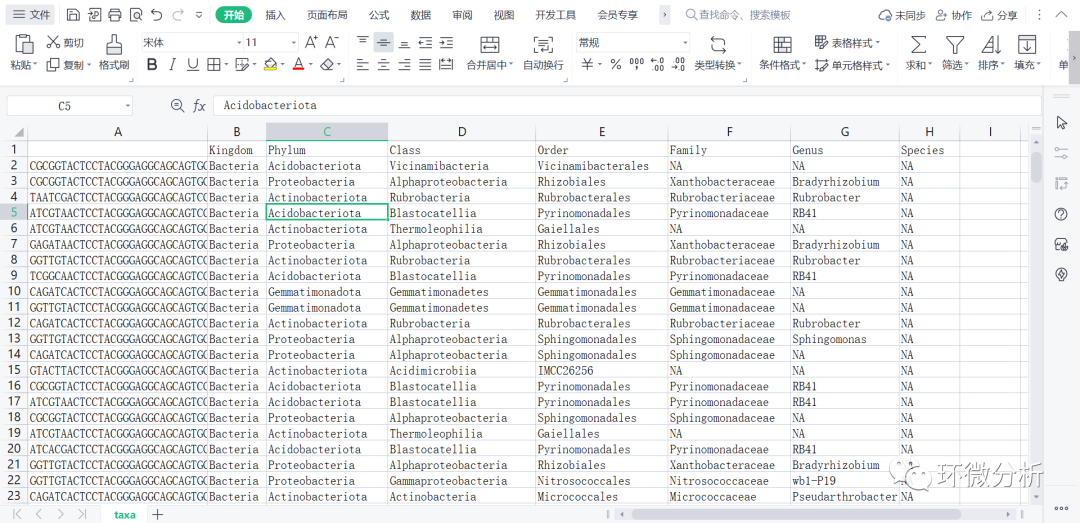
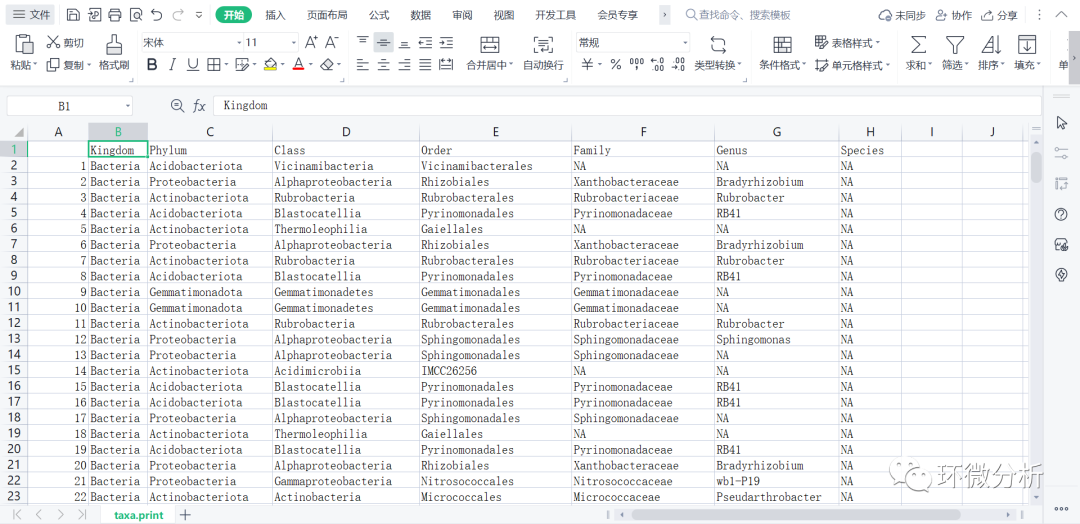
# 另存物种注释变量,去除序列名,只显示物种信息
# Removing sequence rownames for display only
taxa.print <- taxa rownames(taxa.print) <- NULL head(taxa.print)

# 输出taxa.print文件 write.csv(taxa.print,file="C:/Users/ASUS/Desktop/R/rawdata/filtered/taxa.print.CSV",append = FALSE, quote = FALSE , sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
(10)物种注释(方法二)
IdTaxa物种注释分类方法也可通过 DECIPHER Bioconductor包获得。IDTAXA算法的分类性能优于朴素贝叶斯分类器。这里我们包含一段代码区域,允许你使用IdTaxa函数替代assignTaxonomy,并且它更快!。目前较新的经过训练的分类器SILVA_SSU_r138_2019.RData 数据库文件可从 http://DECIPHER.codes/Downloads.html 获得。
# 安装DECIPHER,加载DECIPHER
BiocManager::install("DECIPHER", version = "3.14")
library(DECIPHER)
packageVersion("DECIPHER")
# 转换ASV表为DNAString格式
# Create a DNAStringSet from the ASVs
dna <- DNAStringSet(getSequences(seqtab.nochim))
# 相关数据下载详见DECIPHER教程,并修改为下载目录
# CHANGE TO THE PATH OF YOUR TRAINING SET
load(paste0(path, "/SILVA_SSU_r138_2019.RData"))
# use all processors
ids <- IdTaxa(dna, trainingSet, strand="top", processors=NULL, verbose=FALSE) # ranks of interest ranks <- c("domain", "phylum", "class", "order", "family", "genus", "species")
# Convert the output object of class "Taxa" to a matrix analogous to the output from assignTaxonomy
taxid <- t(sapply(ids, function(x) { m <- match(ranks, x$rank) taxa <- x$taxon[m] taxa[startsWith(taxa, "unclassified_")] <- NA taxa })) colnames(taxid) <- ranks; rownames(taxid) <- getSequences(seqtab.nochim) # Removing sequence rownames for display only taxa.print.DECIPHER <- taxa <- taxid rownames(taxa.print.DECIPHER) <- NULL head(taxa.print.DECIPHER)

# 导出注释结果文件taxa.print.DECIPHER write.csv(taxa.print.DECIPHER,file="C:/Users/ASUS/Desktop/R/rawdata/filtered/taxa.print.DECIPHER.CSV",append = FALSE, quote = FALSE , sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
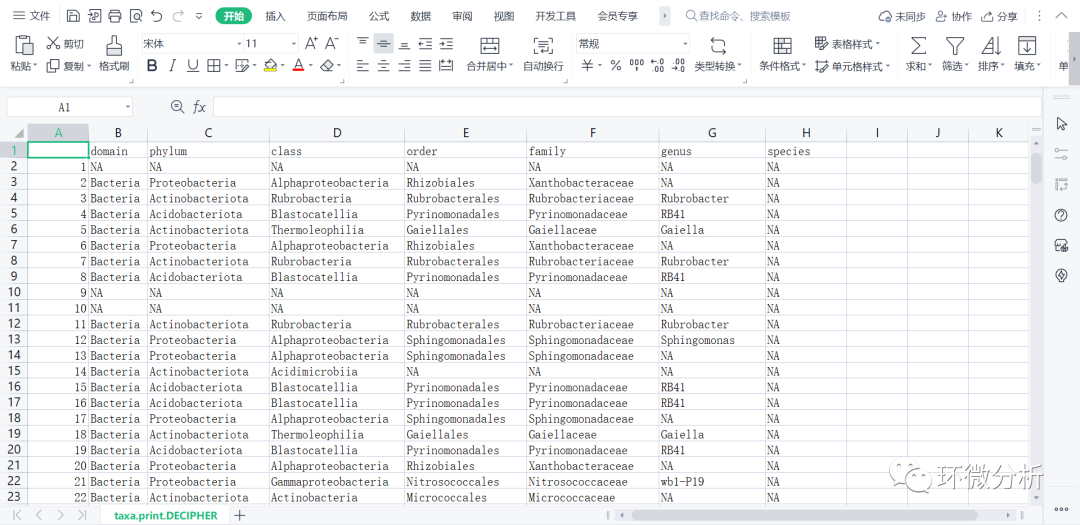
# 注意,上面的行名是NULL,即1、2、3、4……,#使用以下命令输出ASV为行名的注释表 colnames(taxid) <- ranks; rownames(taxid) <- getSequences(seqtab.nochim) taxa.print.DECIPHER.taxa <- taxa <- taxid head(taxa.print.DECIPHER.taxa) write.csv(taxa.print.DECIPHER.taxa,file="C:/Users/ASUS/Desktop/R/rawdata/filtered/taxa.print.DECIPHER.taxa.CSV",append = FALSE, quote = FALSE , sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
提示1:小内存计算机可能运行失败,32G内存+8核笔记本电脑经常出现处理程序终止报错,多试几次即可成功。读入RData后R环境可能崩溃了,R语言可能不擅长处理大数据文件;
提示2:如果您的数据没有被适当注释,例如您的细菌16S序列被分配为大量Eukaryota NA NA NA NA NA, 可能核苷酸序列方向与参考数据库的方向相反。告诉DADA2尝试反向互补方向进行匹配,assignTaxonomy(…,tryRC=TRUE)看看这是否可以修复注释信息。如果使用DECIPHER进行分类,请尝试IdTaxa (…, strand=“both”)。
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!

















 2631
2631

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









