







本节主要讲解使用Mothur软件对扩增子基因序列进行数据库比对,过滤,聚类,去除嵌合体及其处理评估。
01数据与数据库的比对
使用pcr.seqs命令针对感兴趣的区域定制一个数据库,将序列与参考序列比对。参考数据库(silva.bacteria.fasta),序列比对的起始位置为11894、结束位置为25319。为了删除leading and trailing dots,将keepdots设置为false。
| 命令注释:pcr.seqs命令将按照用户自定义的选项修剪已输入序列。 |
屏幕输出:

| 输出结果注释: 1. 在利用mothur的align.seqs模块将tags比对到sivla参考序列时,为了减少计算量,通常会对silva进行区域的筛选;2. v4区域的参考位置是start=11894, end=25319;3. v3-5区域的参考位置是start=6426,end=27654;4. v3-4区域的参考位置是start=6428, end=23444。 |
使用rename.file命令重命名上述输出文件,
| 命令注释:rename.file命令用于对文件重命名,当文件名太长不便使用时使用。 |
屏幕输出:

查看此命令做了什么,屏幕输出:
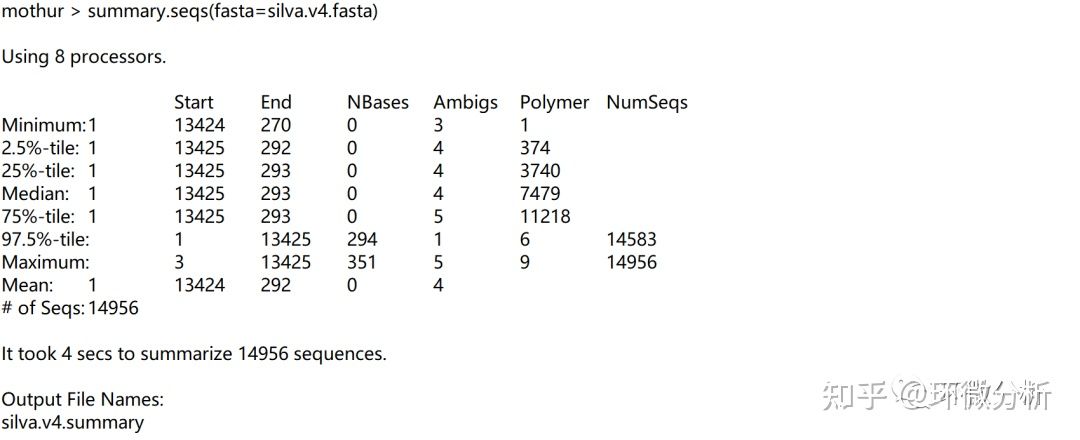
输出文件:silva.v4.summary
现在,有了一个自定义的参考比对,可以与序列比对。好处是,现在不再是50,000列宽,而是13,425列宽,会节省硬盘驱动器一些空间,提高整体对齐质量。
| 命令注释:align.seqs命令用于把用户提供的fasta格式的候选序列文件对齐到用户提供的同样格式的模板序列。 |
屏幕输出:

| 输出结果注释: 1. 使用align.seqs进行对齐,能够根据选择的方法来输入序列之间的比对以及与参考序列的比对。默认情况下,align.seqs将使用8-mers进行k-mer搜索,并将使用Needleman-Wunsch成对对齐方法,对匹配的奖励为+1,对不匹配和间隔的惩罚为-1和-2。2. mothur提供了三种查找模板序列的方法,包括k-mer搜索、blast和suffix tree搜索。k-mer搜索是搜索模板序列的最快和最好的方法。默认是使用k-mers,即在reads上,从第一个碱基开始,一个碱基一个碱基地移动,截取长度为k的DNA序列,称为k-mer,就是长度为k的DNA序列。 |
输出文件:
stability.trim.contigs.good.unique.align
stability.trim.contigs.good.unique.align.report
| 文件注释:QueryName查询名称;QueryLength查询长度;TemplateName模板名称;TemplateLength模板长度;SearchMethod搜索方法;SearchScore搜索得分;AlignmentMethod对齐方法;QueryStart查询开始;QueryEnd查询结束;TemplateStart模板启动;TemplateEnd模板结束;PairwiseAlignmentLength对齐长度;GapsInQuery查询中的间隙;GapsInTemplate模板中的间隙;LongestInsert最长插入;SimBtwnQuery&Template:SimBtwn查询和模版 |
再次运行summary.seqs,屏幕输出:
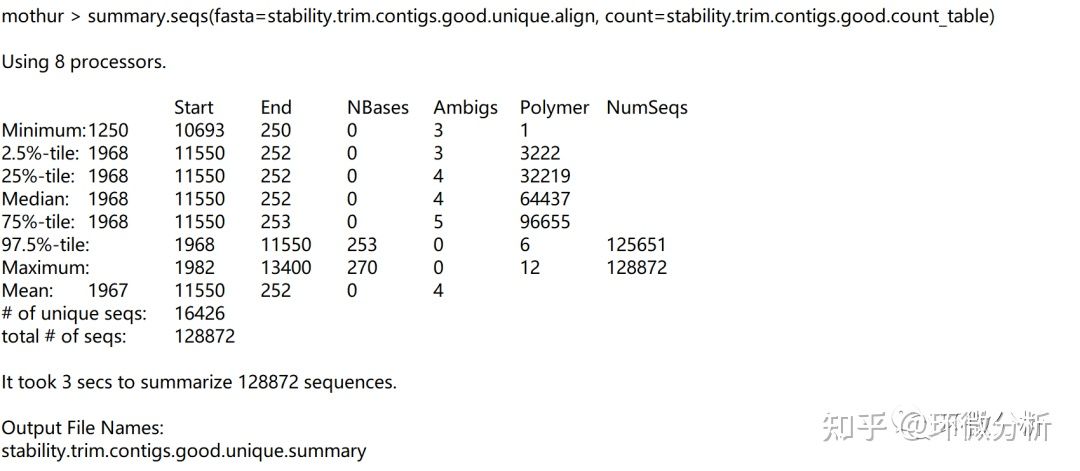
| 输出结果注释:大部分序列从1968位置开始,在11550位置结束。一些序列从1250或1968位置开始,在10693或13400位置结束。偏离位置的情况可能是由于在路线的末端插入或删除。有时会看到序列在同一个位置开始和结束,表明比对非常差,通常是由于非特异性扩增所致。 |
输出文件:stability.trim.contigs.good.unique.summary
为了确保所有内容都overlaps在同一区域上,重新运行screen.seqs以获得在1968位置或其之前开始并在11550位置或之后结束的序列。最大的均聚物长度设置为8,因为数据库中没有任何一个连续9个或更多相同的碱基(这在上面的第一步执行screen.seqs时完成)。需要count表方便更新去除了一定序列的表,而且还使用summary文件,这样就不必再次计算出所有的开始和停止位置。
屏幕输出:

输出文件:
stability.trim.contigs.good.unique.good.summary
stability.trim.contigs.good.unique.good.align
stability.trim.contigs.good.unique.bad.accnos
stability.trim.contigs.good.good.count_table
再次运行summary.seqs,屏幕输出:
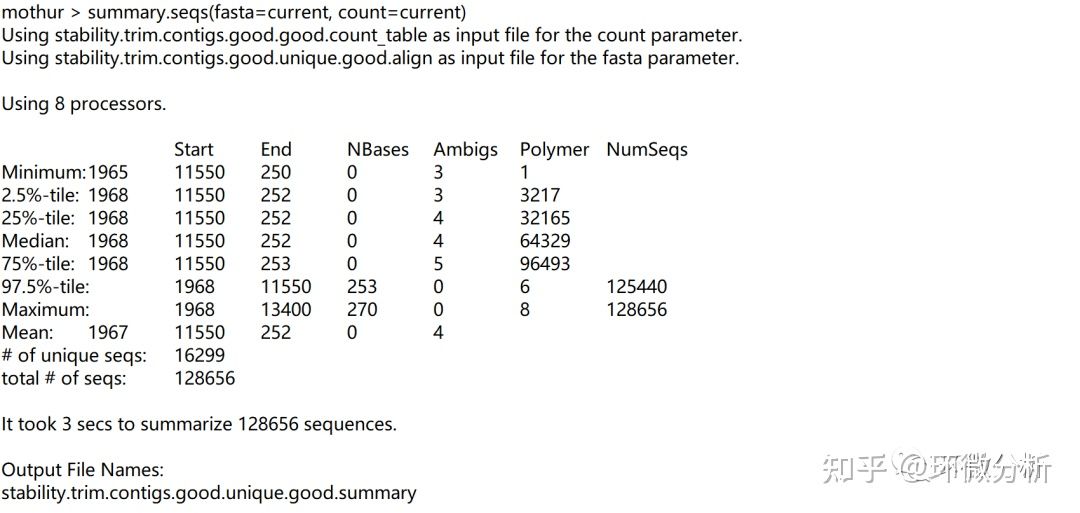
输出文件:stability.trim.contigs.good.unique.good.summary
现在知道序列与相同的比对坐标重叠,但想确保它们仅与该区域重叠。接下来过滤序列以消除两端的overhangs,对齐中有许多列只包含间隙字符(即‘-’)需要取出。用filter.seqs来做这一切:filter.seqs根据用户定义的条件从比对中删除列。例如,针对参考对齐方式(例如,来自RDP,SILVA或greengenes)生成的对齐方式通常具有其中每个字符均为'。'的列或“-”。这些列不包含在计算距离中,因为它们中没有信息。通过删除这些列,可以加快大量距离的计算。
02数据的过滤
使用filter.seqs完成数据过滤工作。
| 命令注释:filter.seqs命令用于根据用户自定义标准从对齐文件中移除列。 |
屏幕输出:

输出文件:
stability.filter
stability.trim.contigs.good.unique.good.filter.fasta
初始对齐是13425列宽,可以使用trump=.删除13049个终端间隔字符,垂直间距字符使用vertical=T.,最终对齐长度为376列。可能已经通过修剪末端在序列中创建了一些冗余,所以重新运行unique.seqs。
屏幕输出:

| 结果注释:确定了3个重复序列,且将其与以前的唯一序列合并 |
输出文件:stability.trim.contigs.good.unique.good.filter.count_table
| 文件注释:第一列代表序列名称 ,第二列代表序列总数为1,其余为各组代表序列个数。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.fasta
03数据的聚类
利用pre.cluster命令对序列进行预聚类,允许序列间最多有2个差异,使序列进一步去噪。这个命令将对序列进行分组拆分,然后按丰度进行选择,从最大丰度到最小丰度,并识别相互之间2 nt以内的序列。如果他们被确定,则他们就会合并。一般接受每100 bp序列有1个差异,
| 命令注释:pre.cluster命令用于实现伪单链接算法(pseudo-singe linkage algorithm),这个算法的主要目的是为了去除那些可能由于焦磷酸测序错误产生的序列。 |
屏幕输出:


输出文件:
stability.trim.contigs.good.unique.good.filter.unique.precluster.fasta
stability.trim.contigs.good.unique.good.filter.unique.precluster.count_table
| 文件注释:第一列代表序列名称,第二列代表序列总数,其余列为每组中代表序列数。 |
现在有6087个唯一序列。接下来使用VSEARCH算法消除嵌合体,chimera.vsearch命令可以实现此功能。此命令将按样本拆分并检查嵌合体。首选方法是使用丰富的序列作为参考。此外,如果一个序列在一个样本中被标记为嵌合,则默认(dereplicate=F)是将其从所有样本中删除。这是我们的做法,
| 命令注释:chimera.vsearch命令用于读取fasta文件和参考文件/fasta格式文件和name/count格式文件,并输出所有潜在的嵌合序列。 |
屏幕输出:


输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.count_table
| 文件注释:统计每个样本每条序列中嵌合体的数量。第一列:代表序列名称,第二列:代表序列总数,后面所有列:每组中各代表序列数量 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.chimeras
stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.accnos
| 文件注释:统计出含有嵌合体的序列 |
04去除嵌合体及“不良品”
使用计数文件运行chimera.vsearch从计数文件中删除嵌合体序列,但仍需要从fasta文件中删除这些序列,运行remove.seqs实现:
| 命令注释:remove.seqs命令利用序列名称和fastq、fasta、name、group、list、count或者是align.report格式的任何一个文件来生成一个不包括列表中某些特定序列的新文件。 |
屏幕输出:

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.fasta
运行summary.seqs,会看到剩下的内容。
屏幕输出:
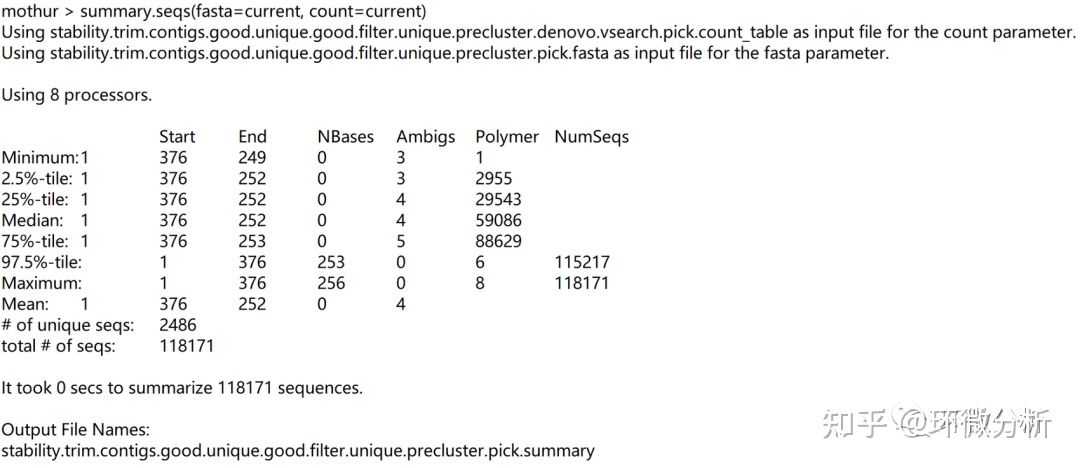
| 结果注释:序列从128656减少到118171,减少8.1%;这是可接受的嵌合体数量。 |
输出文件:
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.summary
作为最后的质量控制步骤,需要确认数据集中是否存在任何“不良品”。继续使用带有classify.seqs命令的贝叶斯分类器对这些序列进行分类。
| 命令注释:classify.seqs命令可使用户使用不同的方法将其筛选出来的序列分配到不同的分类提纲中。 |
屏幕输出:

输出文件:
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.taxonomy
| 文件注释:第一列序列名称,第二列物种注释结果从界到属6个水平。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.tax.summary
| 文件注释: 从界到属6个水平,每一行是一个物种。Taxlevel分类水平;RankID排序名称;taxon分类水平;Root根(起源);Total总计序列数 |
所有内容都已分类,使用remove.lineage命令删除“不良序列”:
| 命令注释:remove.lineage命令用于读取taxonomy文件和taxon文件,并生成一个只包含不在taxon中序列的文件。 |
屏幕输出:

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.taxonomy
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.accnos
stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.count_table
| 输出文件注释:每个样本中代表序列数量表。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.fasta
如果分类器无法将序列分类为一个域,则“unknown”仅作为分类弹出。如果没有使用RDP参考分类法对序列进行分类,还需要自定义谱系的名称。例如,修改后的RDP版本称为mitochondria,“Mitochondria”。如果使用greengenes,它将它们称为“f__mitochondria ”。如果正在使用greengenes(或SILVA或其他),则需要适当更改这些名称。
现在,要创建反映这些删除的更新的taxonomy.summary文件,使用summary.tax命令。
| 命令注释:summary.tax命令用于读取taxonomy文件和选择性的name或group文件,并总结taxonomy信息。 |
屏幕输出:

创建一个pick.tax.summary文件,其中删除了“不良序列”。
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.tax.summary
名词解释
嵌合体(Chimera):即嵌合基因。即两个基因共用一段序列,这两个基因称为嵌合基因。
嵌合体序列:由来自两条或者多条模板相连的序列组成。细菌多样性研究中,产生嵌合体的主要原因是在16S扩增过程中不完全的模板延伸造成的,在一个循环中由一个序列连接到另一条序列的部分区域上。
贝叶斯分类器(Bayes classifier):各种分类器中分类错误概率最小或者在预先给定代价的情况下平均风险最小的分类器。分类原理:通过某对象的先验概率,利用贝叶斯公式计算出其后验概率,即该对象属于某一类的概率,选择具有最大后验概率的类作为该对象所属的类。
Chloroplast:叶绿体
Mitochondrian:线粒体
Archaea:古菌
Eukaryota:真核生物
bp:base pair碱基对
nt:Nucleotide核苷酸
Dereplicate:去复制
05数据处理评估
使用get.groups命令从“Mock”样本中提取序列:
| 命令注释:get.group命令提供用于当前存储的多样本otu数据的分组列表。 |
屏幕输出:

| 输出结果注释:Mock样本中有64个唯一序列,总共4048个序列。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta
使用seq.error命令来测量错误率,
| 命令注释:seq.error命令用于读取fasta文件并基于参考文件寻找其中序列中的错误。 |
屏幕输出:

| 结果注释:结果显示错误率是0.0065%。 |
输出文件:
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.summary
| 文件注释:该文件统计详细统计了单条序列错误情况 Query问题序列;reference参考序列;Substitutions替换;Ambig模糊碱基;Matches匹配;Mismatches不匹配;Total总计;Error错误率 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.seq
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.chimera
| 文件注释:Querynam问题序列名称;Bestref,bestreference最佳参考;bestSequenceMismatch与最佳序列不匹配;minMismatchToChimera嵌合体检 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.seq.forward
| 文件注释:正向错误序列 Position位置;totalseqs序列总计;Match or mismatch能比对上的碱基数(完全匹配或错配);Substitution取代率;Insertion该位置插入碱基数;Deletion该位置缺失碱基数;ambiguous模糊碱 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.seq.reverse
| 文件注释:反向错误序列。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.count
| 文件注释:第一列是错误数量,第二列序列数目。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.matrix
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.error.ref
名词解释
错误率:与参考不匹配的总和/查询中的碱基总和
06数据后处理
将序列聚类到OTU中,以查看有多少个伪OTU。
| 命令注释:dist.seqs命令用于计算已对齐的DNA序列未校正的成对距离(双尾距离,pairwise distanc),生成列或叶格式的距离矩阵。Cutoff=0.03意思为不保存任何大于0.03的距离。 |

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.dist
| 文件注释:前两列为序列名称,第三列为两个序列的距离。 |
| 命令注释:cluster命令用于将已对齐的序列聚类为OTUs,前提是在mothur中输入距离矩阵。 |
屏幕输出:
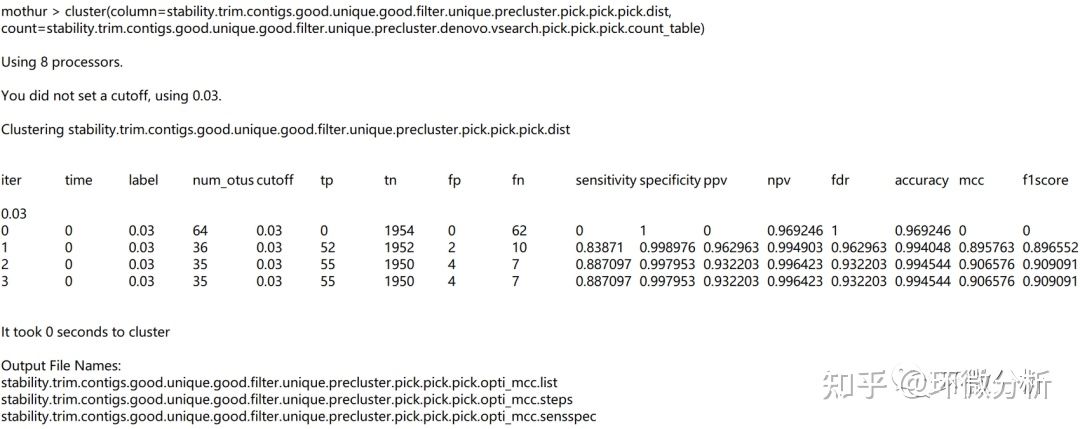
输出文件:
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.list
| 文件注释:文件共三列,含义:0.03水平下,OTUs的数量,每个OTU中的序列 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.steps
| 文件注释: Matthews相关系数(mcc);灵敏度(sens);特异性(spec);真阳性+真阴性(tp tn);假阳性+假阴性(fp fn);真阳性(tp);真阴性(tn);假阳性(fp);假阴性(fn);f1分数(f1score);准确性(accuracy);阳性预测值(ppv);阴性预测值(npv);错误发现率(fdr);默认值= mcc |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.sensspec
| 命令注释:make.shared命令用于读取list和group、biom文件并为每个组创建shared和rebund文件。 |
屏幕输出:

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.shared
| 文件注释:该文件内容为每组样本各OTU的数量。 |
| 命令注释:rarefaction.single命令通过无替代的二次抽样方法生成每个样本的稀释曲线。 |
屏幕输出:

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.groups.rarefaction。
这部分要完成两件事——给OTU、ASV分配序列以及进行系统发育工作。首先使用remove.group命令从数据集中删除Mock样本:
| 命令注释:remove.groups命令用于从fasta、name、group、list、taxonomy和shared格式文件中的某个组或组群中移除序列。 |
屏幕输出:

输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.denovo.vsearch.pick.pick.pick.count_table
| 文件注释:第一列是代表序列名称,第二列是代表序列总数,其余为每组代表序列数目。 |
输出文件:stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.fasta
stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pds.wang.pick.pick.taxonomy
| 文件注释:第一列是序列名称,第二列是从界到属水平的物种分类情况。 |
本文提供所有输出文件,百度网盘下载链接:
https://pan.baidu.com/s/19IRtyT-Nb_14O8rEAnk6fg
提取码:1234
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
Mothur3进阶_Mothur扩增子基因序列处理_数据比对、聚类及其处理评估
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!















 314
314











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









