







网络分析(Network analysis)是指通过连接法,寻找变量之间的联系,以网络图或者连接模型(Connection model)来展示数据的内部结构,从而简化复杂系统并提取有用信息的一种定量分析方式。网络分析工具和网络思维被广泛用于数学、社会科学、计算机科学和环境微生物学等领域,主要用于探索一个或多个系统中的实体之间的相互作用。例如在微生物生态研究中,既可以通过分析一个物种群落数据集来展现物种间的共现模式(co-occurance pattern),也可以结合物种群落数据集与环境因子数据集来分析环境因子与生境物种的相互作用关系。网络分析能够使用相关性矩阵分析不同来源的数据集(PS.微生物数据与理化数据),是一种比较自由的分析方法。 一、网络分析在微生物研究领域的应用
网络分析是一种微生物群落结构研究的主流方法,其整合了多种类型的信息,也可以代表系统水平的行为。近年来,人们逐渐开始使用微生物基因序列数据来探索环境样品中共存的微生物类群之间的直接或间接相互作用,网络分析被大量应用于探索复杂多样群落中微生物类群之间的相互作用(即共现模式)。通过对大量环境样品中的微生物类群信息进行网络分析,帮助研究人员预测枢纽物种、优势物种、稀有物种和其他物种之间的联系与相互作用,进而辅助鉴定微生物的演替状态和生态位;此外,结合微生物丰度数据与环境因子数据,可以预测微生物物种与环境因子的相互作用关系。理想情况下,样本集应覆盖环境条件下的空间或时间梯度,以便在分类单元丰度上有足够的变异性来解释共现模式。 二、微生物网络(分析)的作用
微生物网络是由节点和边组成的生态系统在时间或空间状态上的快照(体现),点线的形式可以包含非常丰富的信息。节点通常代表微生物,但也可以代表其他被研究的变量,如溶解氧或总氮等环境因子。边表示节点之间的具有统计学意义的关联关系,连接到节点的边数称为节点的度。网络分析作为二代测序大数据集分析所需的新方法,超越了对自然微生物群落组成和多样性的基本清单描述。网络分析能够探究微生物类群之间和物种与环境因子之间潜在的相互作用机理,有助于确定未培养微生物所占据的功能角色或环境生态位;对重要类群共现模式的网络分析有助于破译跨空间或时间梯度的复杂微生物群落结构。随着微生物群落序列数据的不断积累,我们也开始探索将微生物群落分析扩展到探索代表大多数基于序列的微生物群落分析的α和β多样性模式之外,即使用网络分析在大型复杂数据集中探索和识别新的模式,帮助研究者探索和识别使用标准α/β多样性分析难以发现的模式。 三、常用的网络分析可视化软件与步骤
常用的网络分析可视化软件有Cytoscape与Gephi,二者具备图形化交互式操作界面与完善的图衡量指标、图布局方式,具有无需编程基础、开源免费、操作界面友好、导入数据方便、具有中文语言、兼容Mac与Windows等优点,是两款易学、易用又强大的工具!也可以使用R语言或者绘图网站在线绘图,但是R语言需要一定的语言基础,在数据导入、图形调整与美化等方面比较困难,在线绘图则存在绘图参数设置少,结果输出格式选择少,不利于后续美化。
本文使用Cytoscape与Gephi软件对物种数据与环境因子数据进行相关性网络分析,其主要步骤是:①计算不同研究变量之间的相关系数矩阵,根据相关系数矩阵确定变量之间的相关关系,具体相关性分析原理请参考相关性分析原理与实操;②将相关性分析结果可视化为网络结构图,根据个人需求调整网络图的节点与边的内容与格式,即是以节点与边的形式呈现出网络系统内所有个体间的相关关系。
———— Cytoscape
一、软件安装
(1) 进入cytoscape官网下载界面https://cytoscape.org/download.html,点击“Download3.9.0”。根据自己的电脑配置进行选择下载,小编选择下载64bit版本;
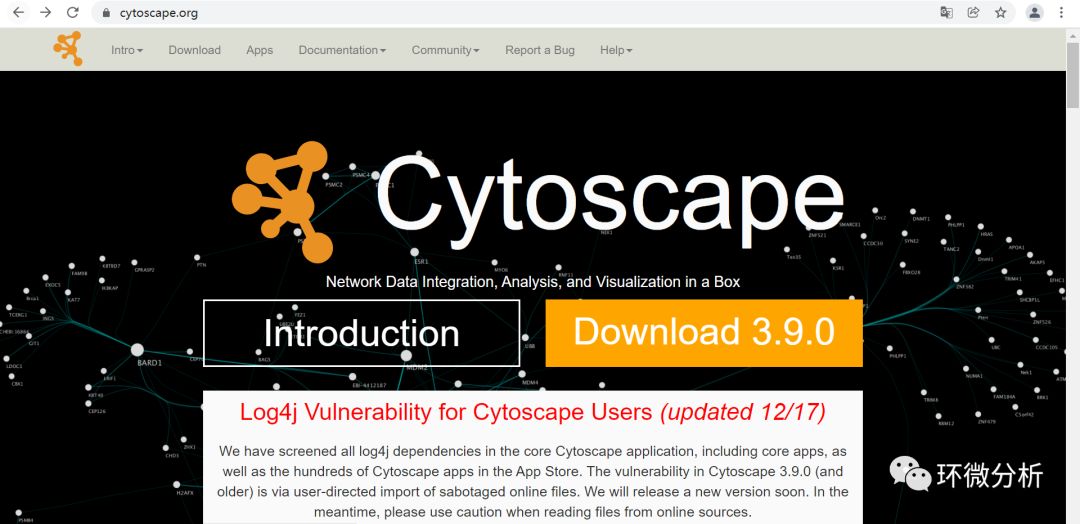
(2) 双击打开下载的文件Cytoscape_3_9_0_windows_64bit.exe,提示安装4j Wizard,选择“Download”开始安装;
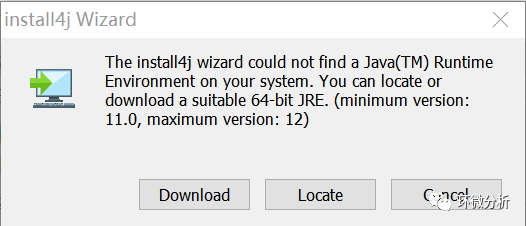
(3) 4j Wizard安装完成,弹出安装提示窗口,点击“Next”;
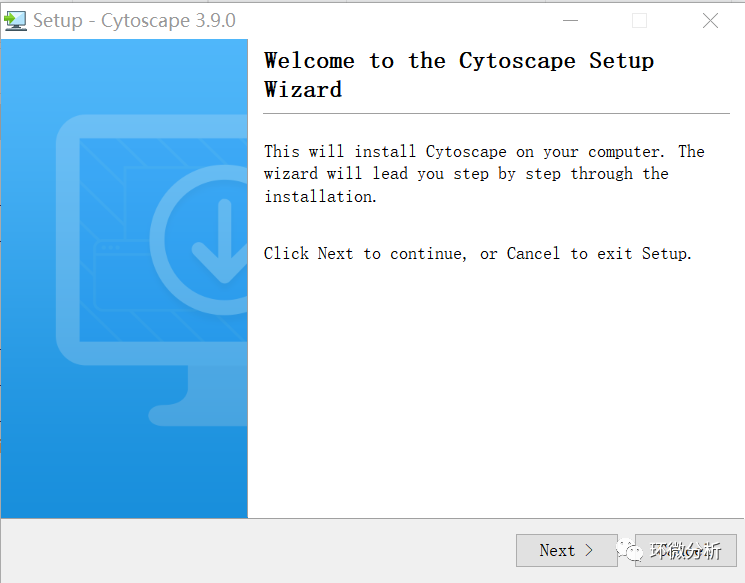
(4) 选择“I accept the agreement”,点击“Next”;
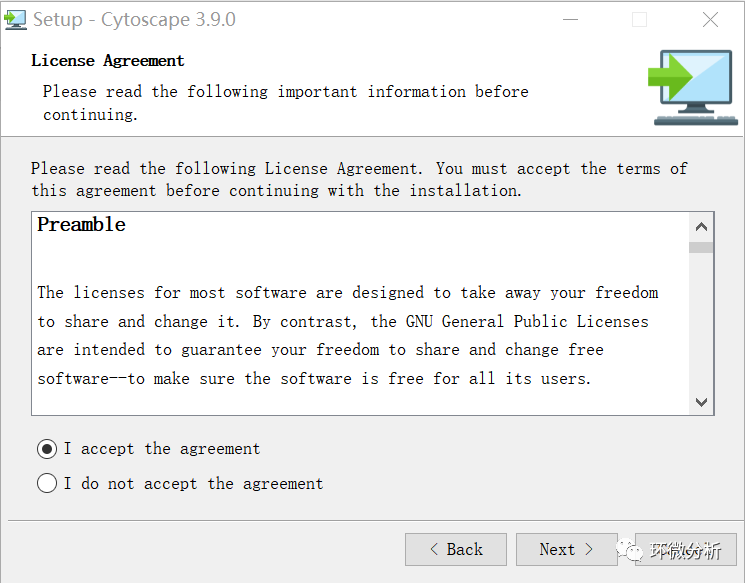
(5) 选择合适的安装位置,点击“Next”;
(6) 勾选“Create a desktop icon”,点击“Next”;
(7) 勾选“Cytoscape Session File(*.cys)”,点击“Next”;
(8) 勾选“Allow collection of generic usage information”,点击“Next”;

(9) 点击“Next”即可安装;
(10) 安装成功,弹出提示框,现在就可以在桌面上找到Cytoscape快捷方式的图标,双击打开即可使用;
提示:Cytoscape的安装需要有Java的环境。如果电脑事先没有安装Java环境,在安装Cytoscape过程中会提醒安装,选择“accept”即可。或者进入Java官网下载界面https://www.java.com/zh-CN/download/获取Java安装程序。
二、数据处理
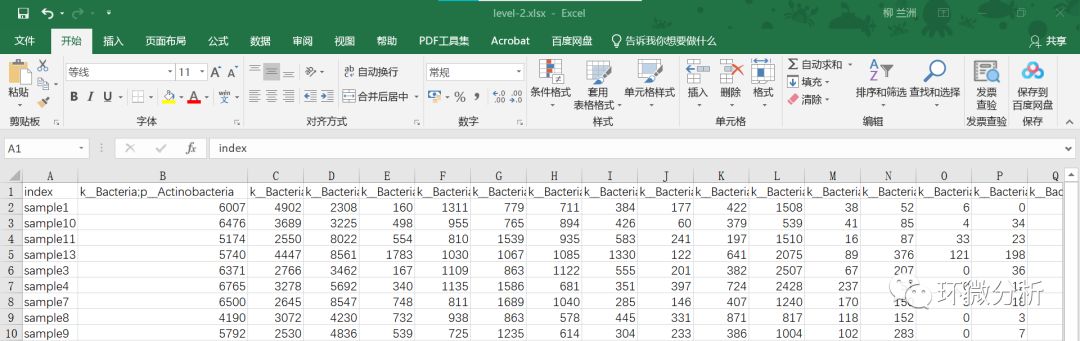
(1) 打开绘制堆叠柱状图所用的数据文件level-2.xlsx,原始数据如下图所示。关注“环微分析”公众号,后台回复“网络分析”即可获取示例数据;
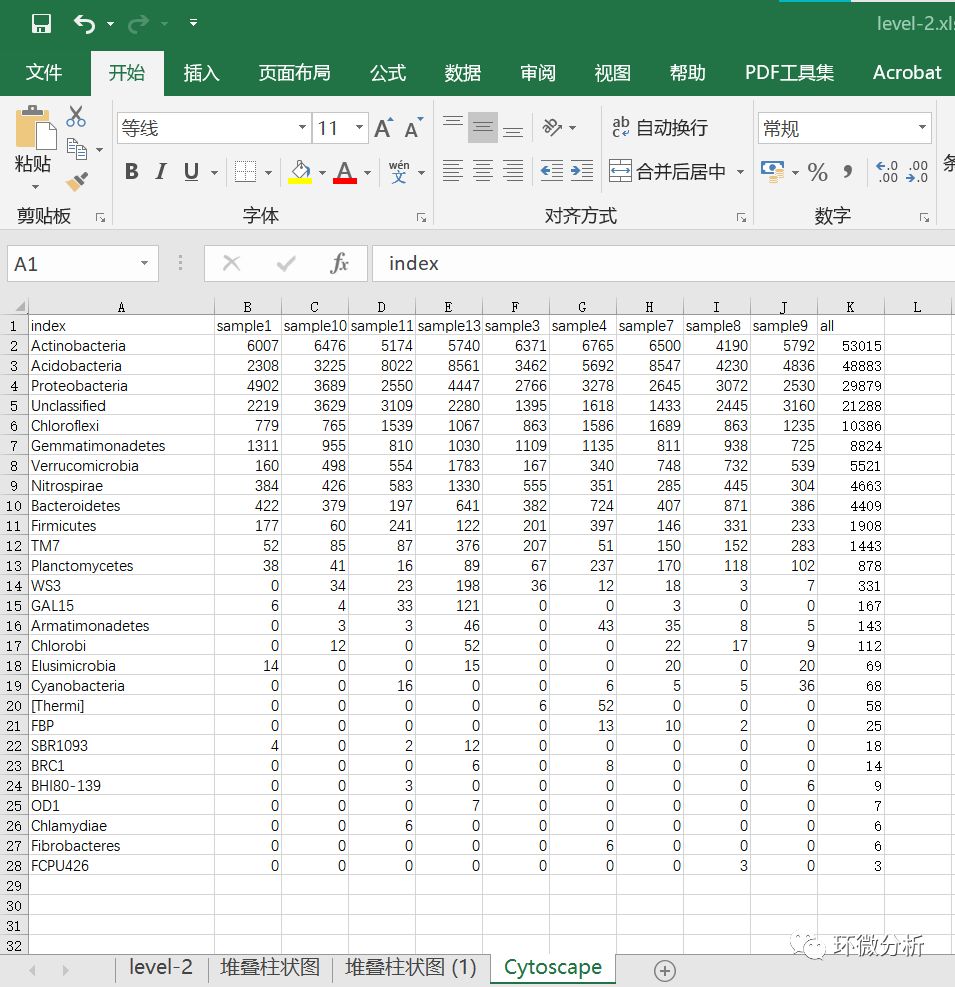
(2) 在工作簿中新建工作表并重命名为“Cytoscape”,复制level-2工作表数据内容,转置粘贴到Cytoscape工作表,对A列内容以分隔符“;”分列,保留门水平列,合并Unclassified,删除A列门名称内容的“p__”,并在K列对9个样品加和数据进行排序。处理结果如下:
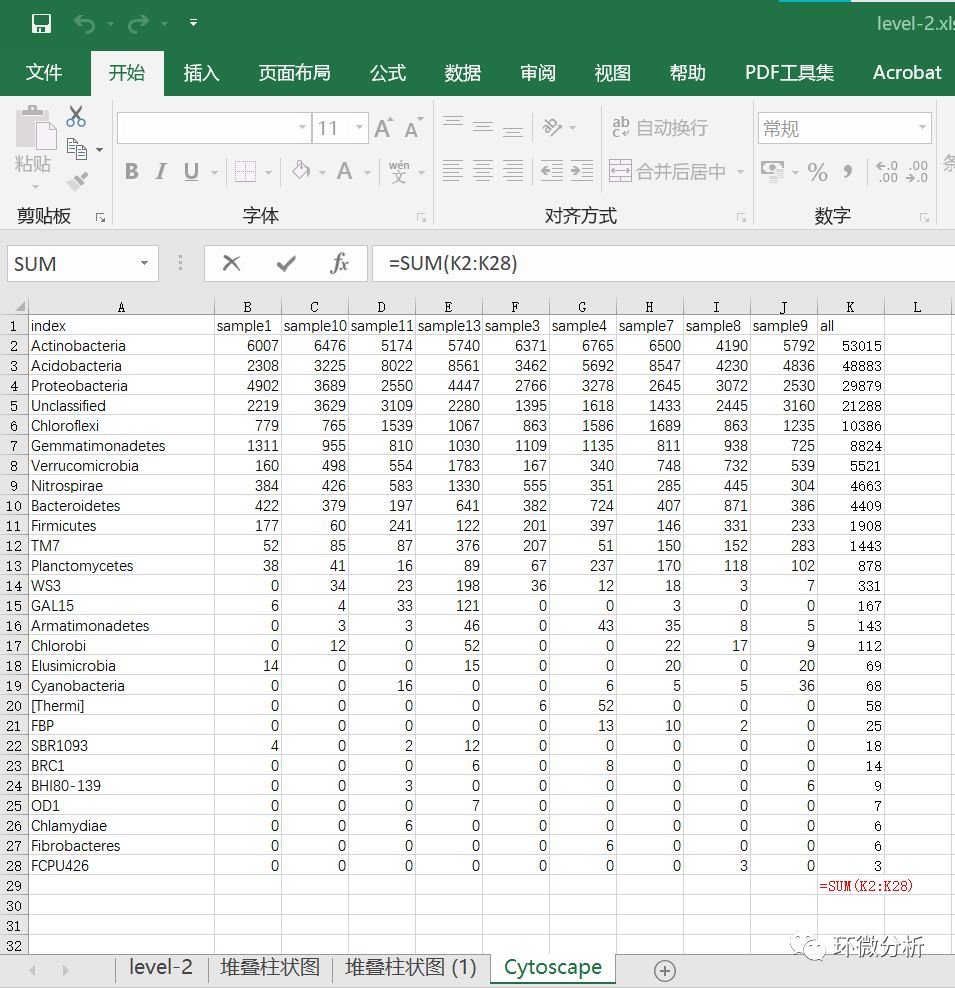
(3) 在K29单元格使用公式“=SUM(K2:K28)”计算9个样本总量;
(4) 在L2单元格使用公式“=K2/$K$29”计算不同菌门的在九个样本的平均占比;
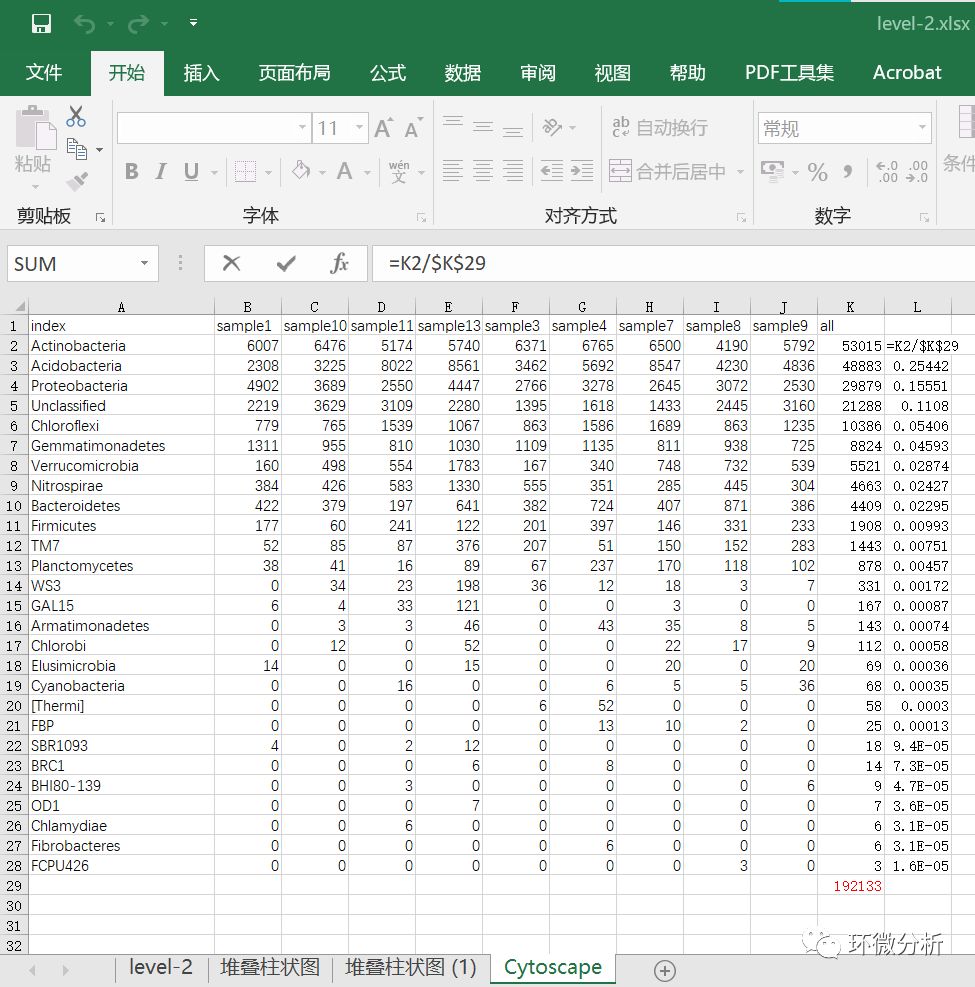
(5) 我们认为平均占比(平均相对丰度)大于1%的菌门属于优势菌门,由上图可以看出2-10列是优势菌门,所以将11-28列合并为Others。结果如下:
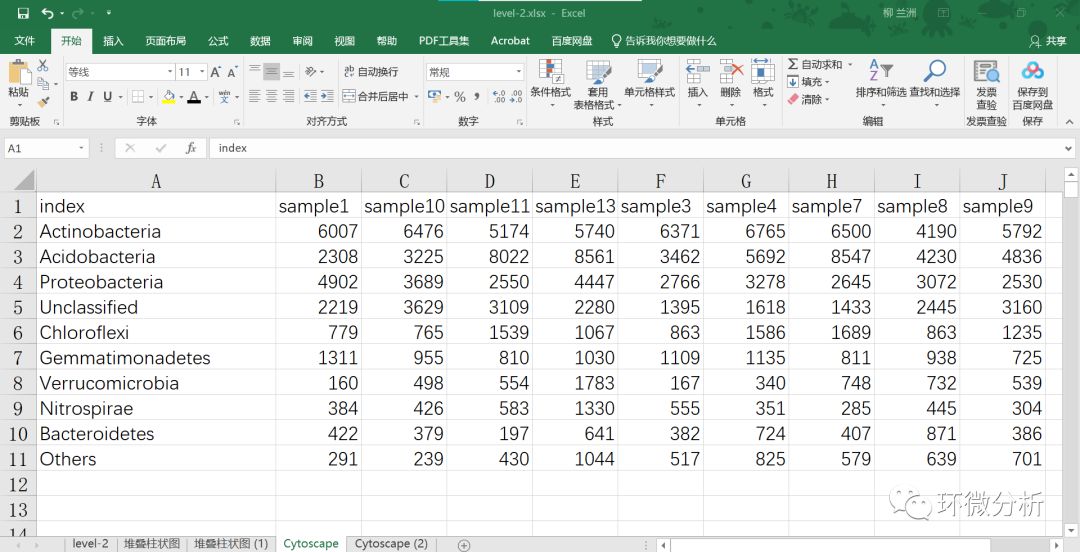
(6) 对数据进行转置粘贴,并对转置数据的A列样本名称进行排序调整;
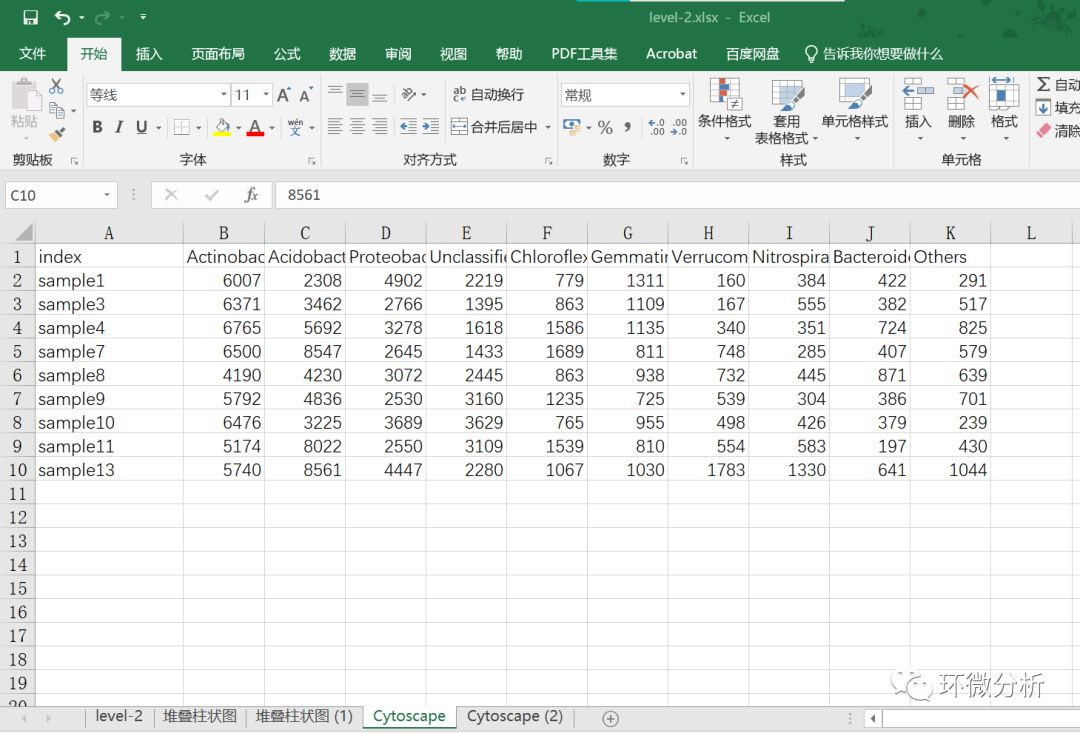
(7) 查看存放在理化.xlsx工作簿下的理化工作表中理化数据;
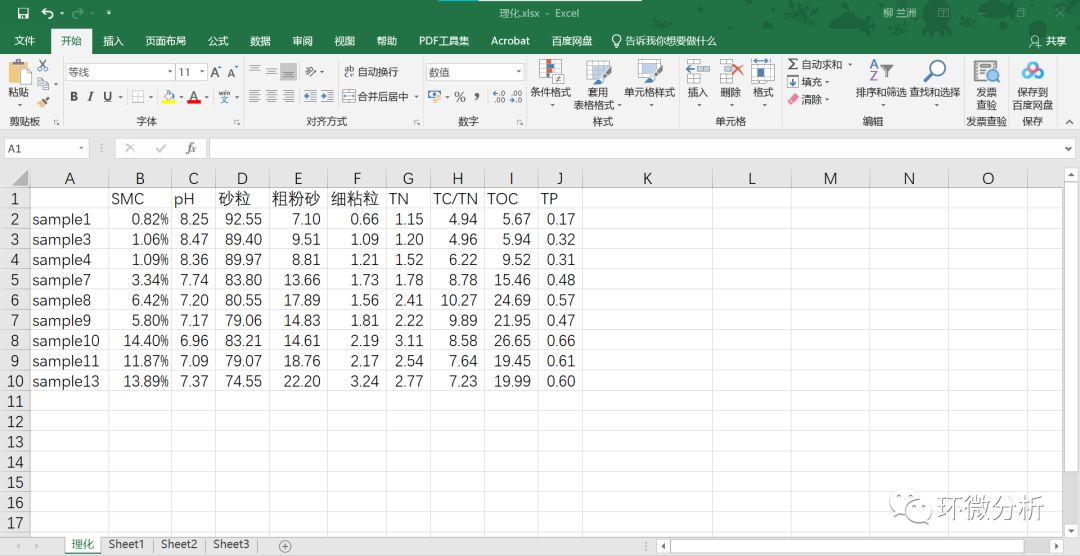
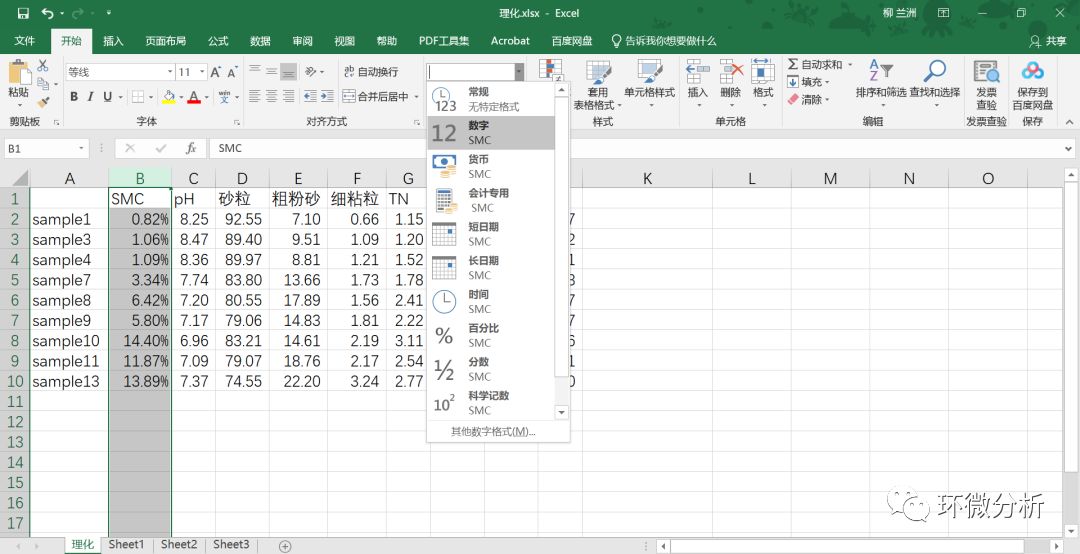
(8) 将B列的数据格式修改成“数字”,并保留4位小数;
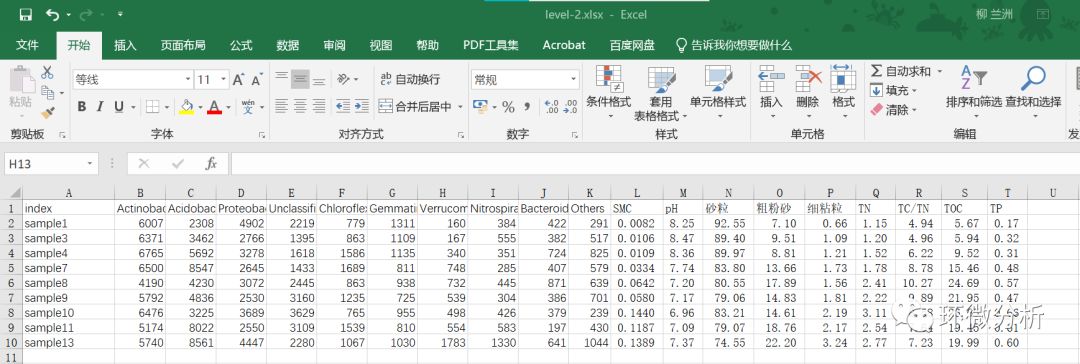
(9) 将理化数据B-J列,1-10行对应粘贴到level-2.xlsx的Cytoscape工作表L列后面,注意一定要A列样本名称对应;
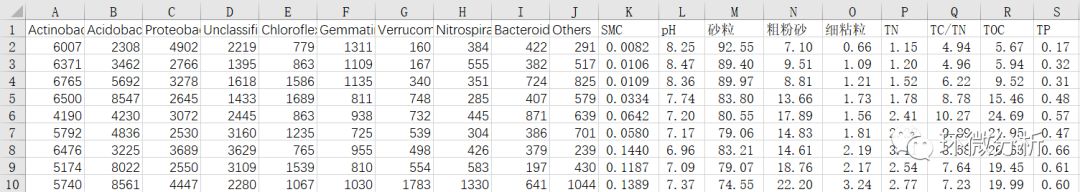
(10) 此时,可以删除A列(样本名称列),因为微生物物种信息已经和理化数据对应好了,可以进行相关性分析了;
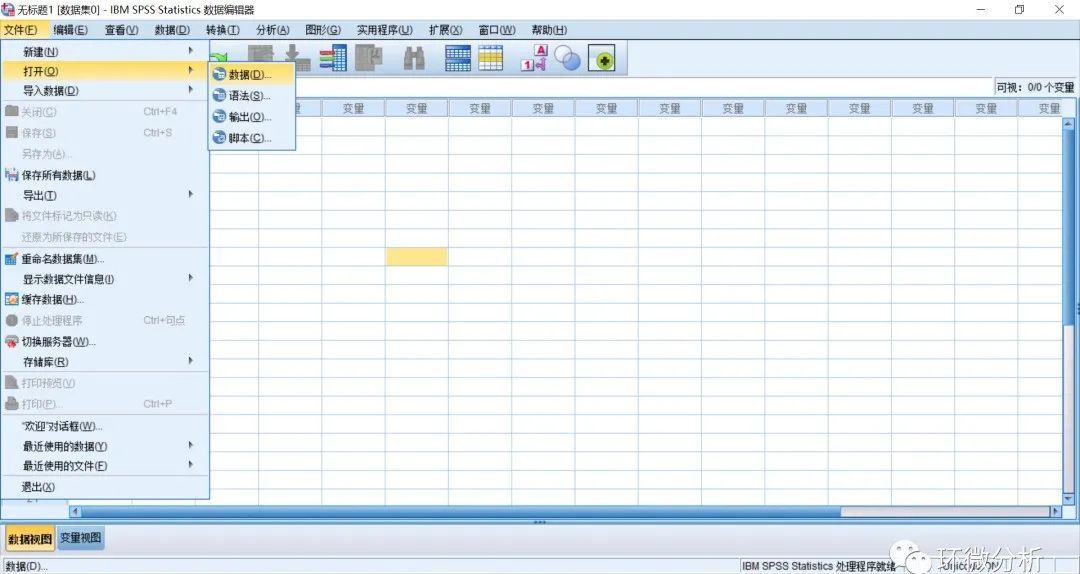
(11) 打开SPSS26,选择“文件”>“打开”>“数据”;
(12) 弹出对话框,文件类型选择“Excel”,文件名选择“level-2.xlsx”,点击打开;

(13) 选择工作表Cytoscape,点击“确定”完成导入;
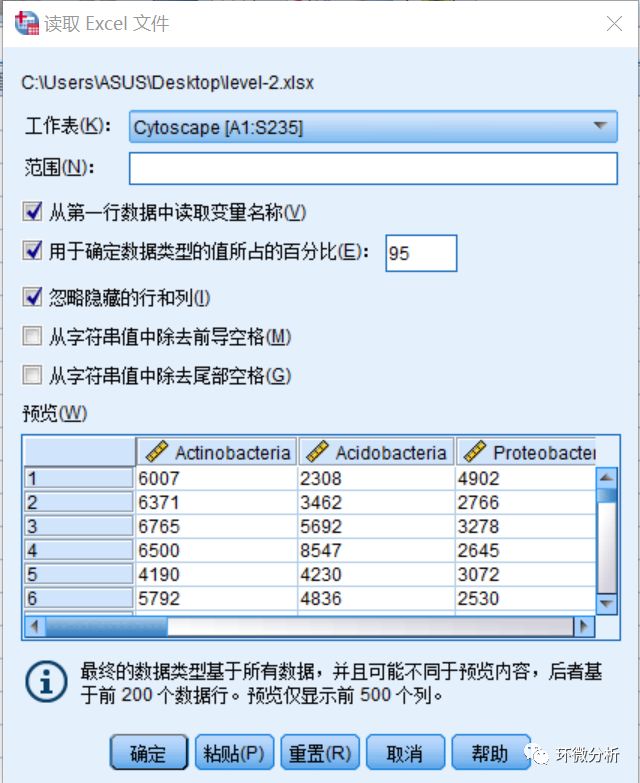
(14) 导入结果如下图所示:
(15) 全选数据,选择“分析”>“描述统计”>“探索”;
(16) 将所有变量全选放入“因变量列表”框中,点击“统计”按钮,在统计框中勾选“描述”,点击“继续”;
(17) 点击“图”按钮,在图框中勾选“含检验正态图”,点击“继续”;然后在“探索”界面,点击 “确定”开始计算;
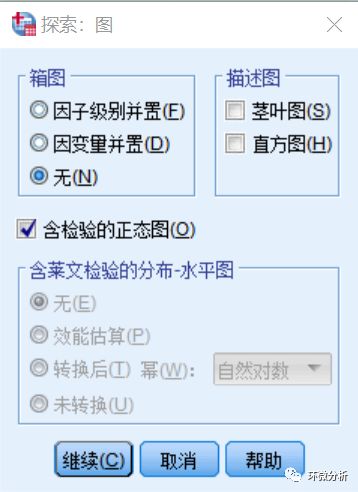
(18) 计算完成,下来输出文档至“正态性检验”部分,其中有柯尔莫戈洛夫-斯米诺夫检验(K-S)与夏皮洛-威尔克检验(S-W)结果。一般来说,当分析大于50行的大样本数据时,我们倾向于采用K-S检验得到的正态性检验结果;当分析小于50行的小样本数据时,我们倾向于采用S-W检验得到的正态性检验结果;当数据量大于5000行时,SPSS只会显示K-S检验这一种检验方法。所以,这里我们示例采用的是夏皮洛-威尔克检验结果。查看S-W检验“显著性”列,显著性检验P小于0.05可视为不显著,即不符合正态分布,P大于0.05可视为符合正态分布。可见,Verrucomicrobia与Nitrospirae变量不符合正态分布;
(19) 返回主页面,全选数据,选择“分析”>“相关”>“双变量”;
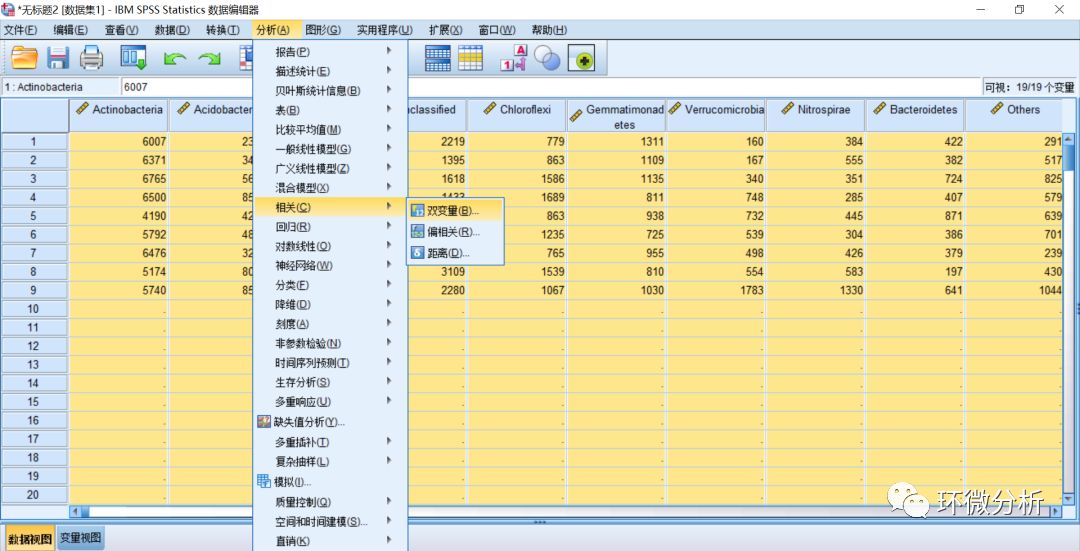
(20) 将所有变量全部放入变量框,相关系数选择“席皮尔曼”,显著性检验选择“双尾”,点击“确定”;在计算变量之间的相关系数时,我们最常使用的是两种相关系数是皮尔逊(Pearson)和席皮尔曼(Spearman)。两种算法的区别在于Pearson要求变量均符合正态分布,而当其中一个变量不符合正态分布时,就需要使用Spearman。本示例中的微生物数据Verrucomicrobia与Nitrospirae变量不符合正态分布,因此使用Spearman算法。其实,大数据集中往往存在不符合正态分布的数据,所以Spearman算法较为常用;
(21) 计算结果如下图所示:
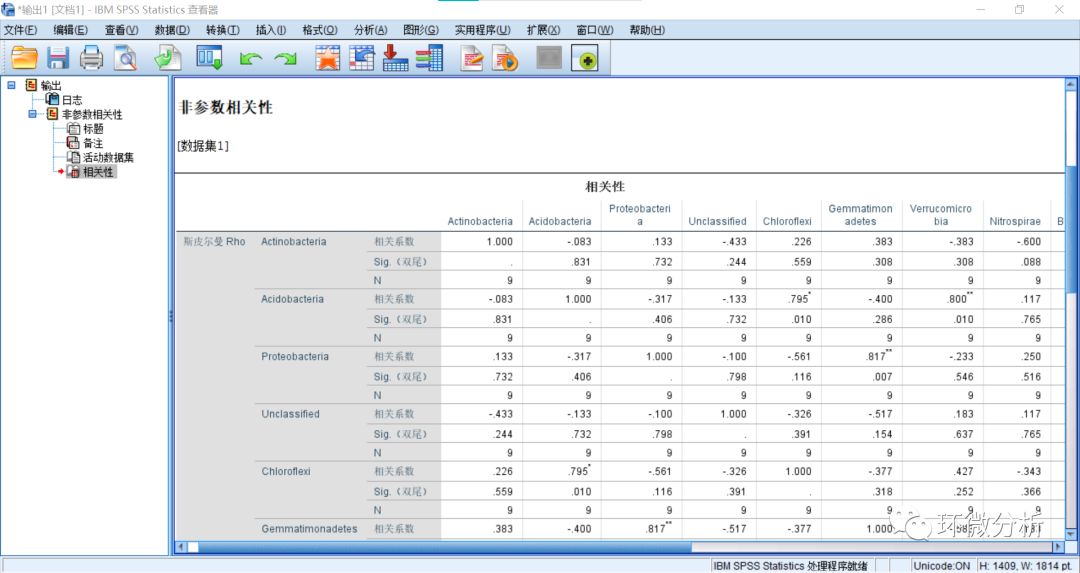
(22) 双击激活输出的“相关性表格”;
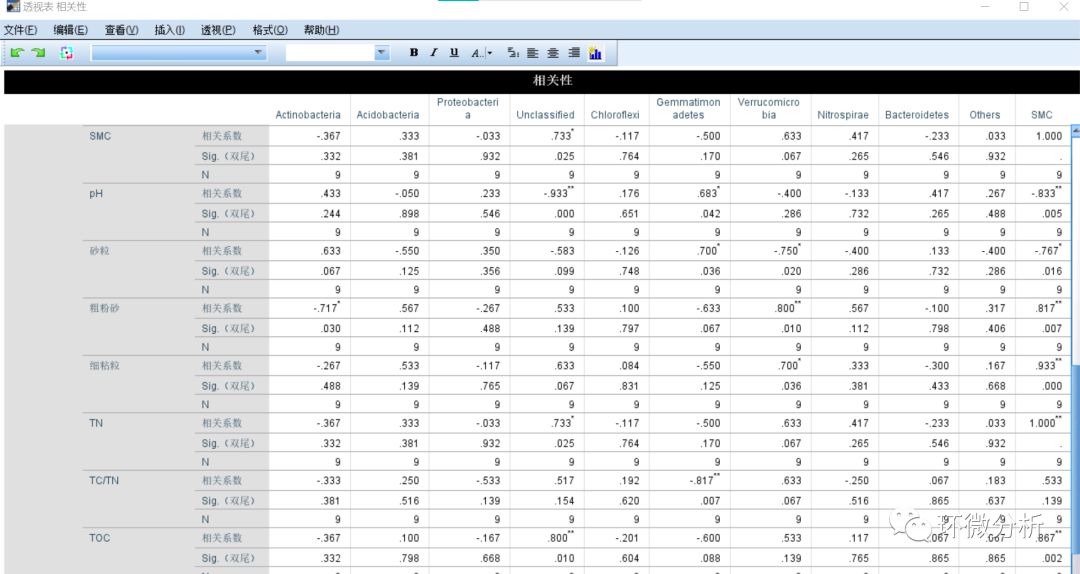
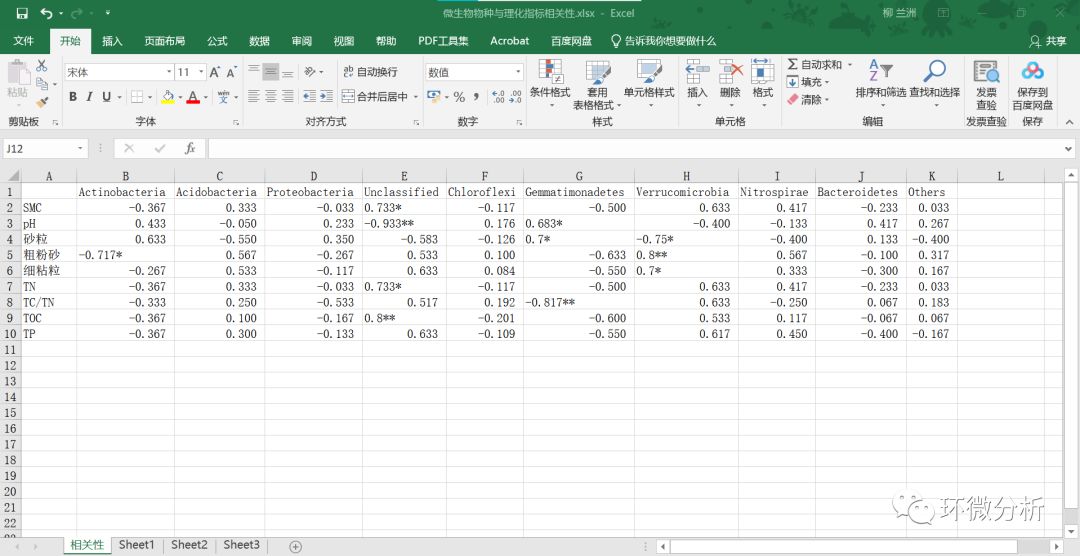
(23) 将微生物门水平的物种数据与理化指标数据的相关系数整合到excel中,一对一复制黏贴即可,注意需要手动补充SPSS中不体现的个位数“0”。整理结果保存为微生物物种与理化指标相关性.xlsx 的“相关性”工作表,数据如下图所示:
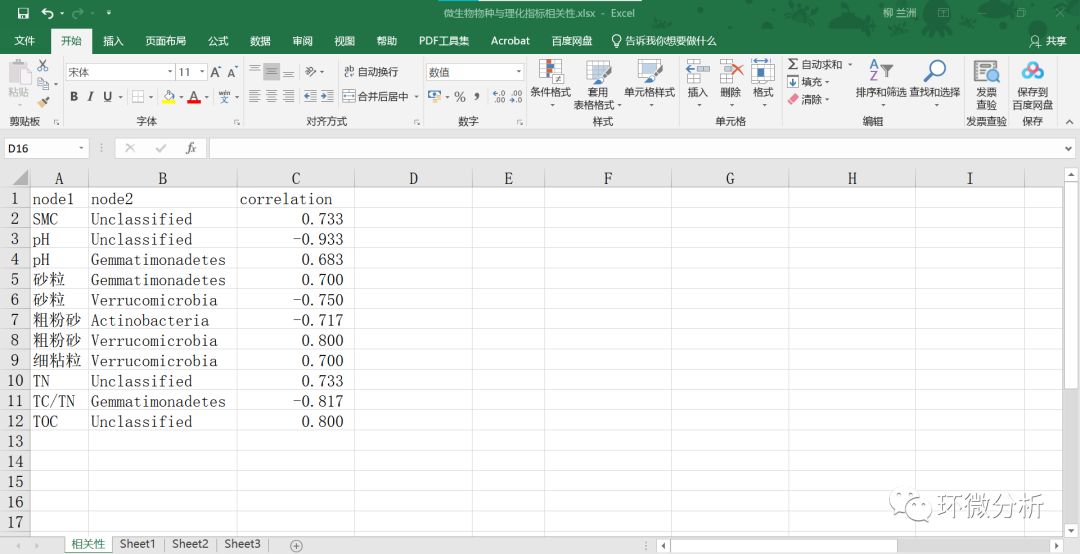
(24) 仅保留具有显著性标记“*”和“**”的数据对,比如保留E2单元格,其对应微生物Unclassified和环境因子SMC的相关性。按对应关系调整成三列,A列为理化指标数据,B为微生物门水平物种数据,C列为同行A列-B列的相关系数,保存。
三、作图流程
(1) 打开Cytoscape软件,进入界面。选择“File”>“Import”>“Network from file”;
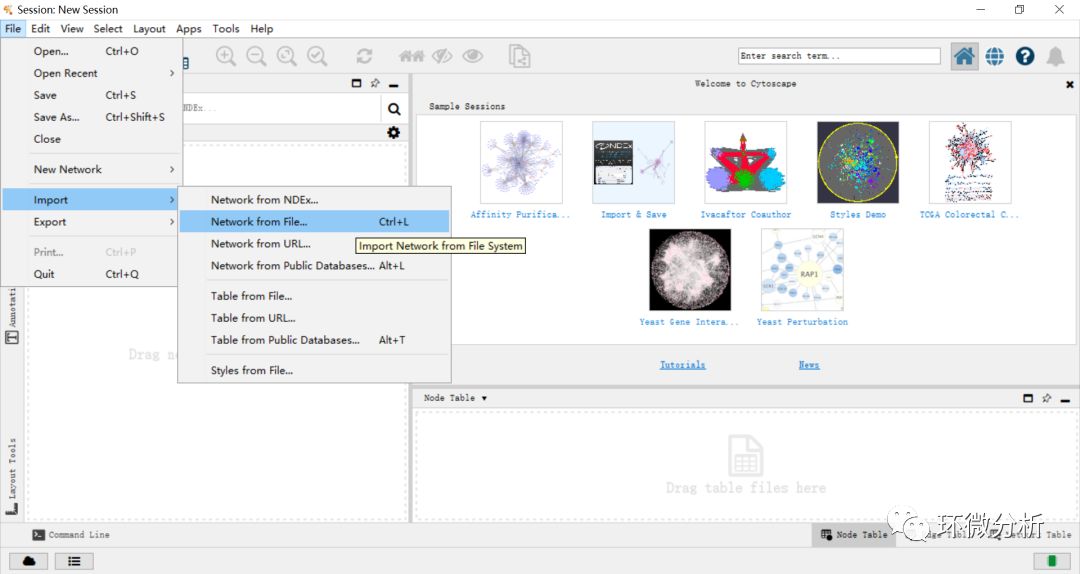
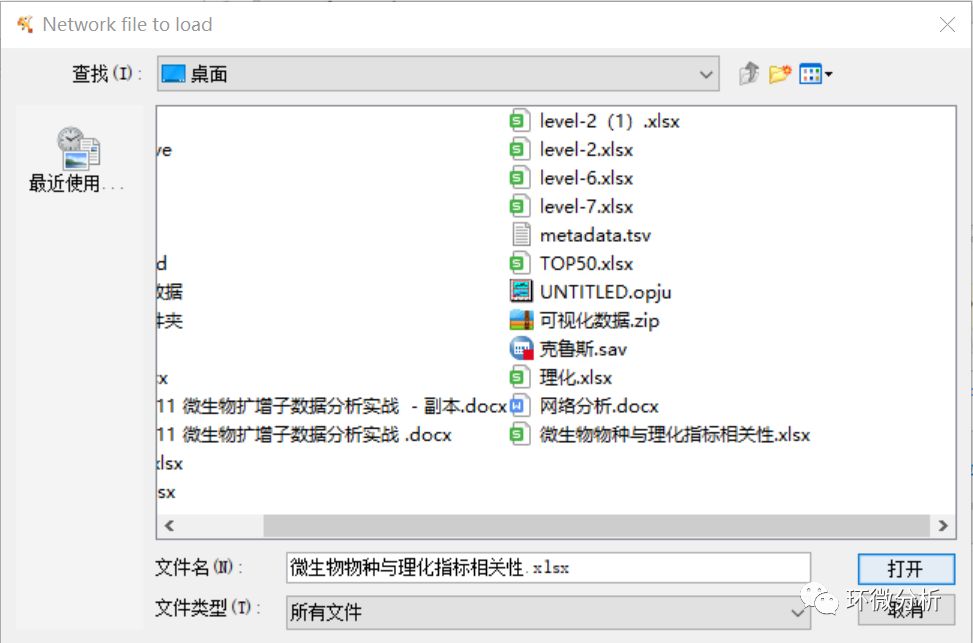
(2) 弹出文件加载框,选择微生物物种与理化指标相关性.xlsx,点击“打开”;
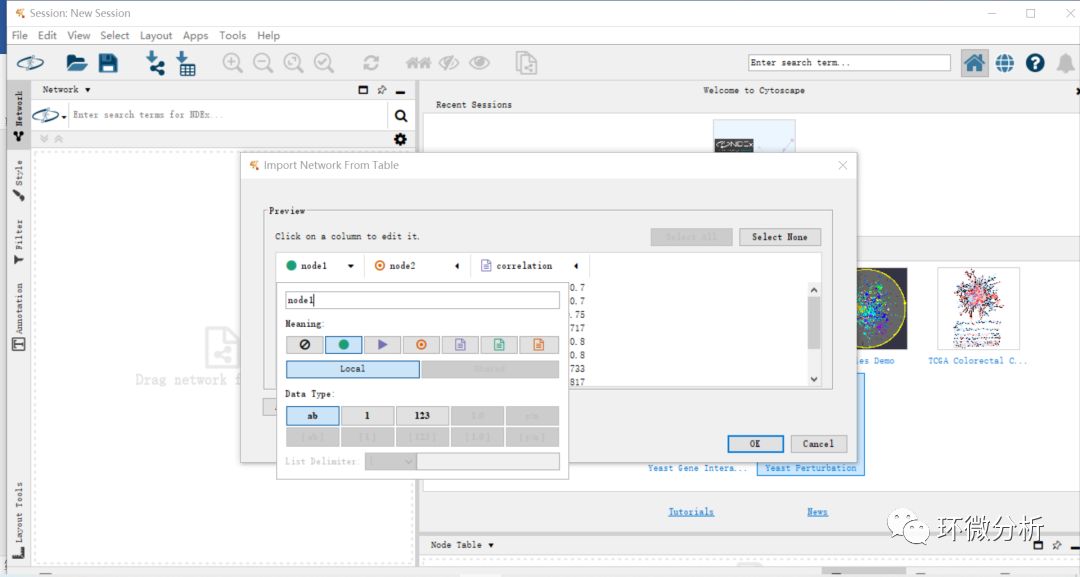
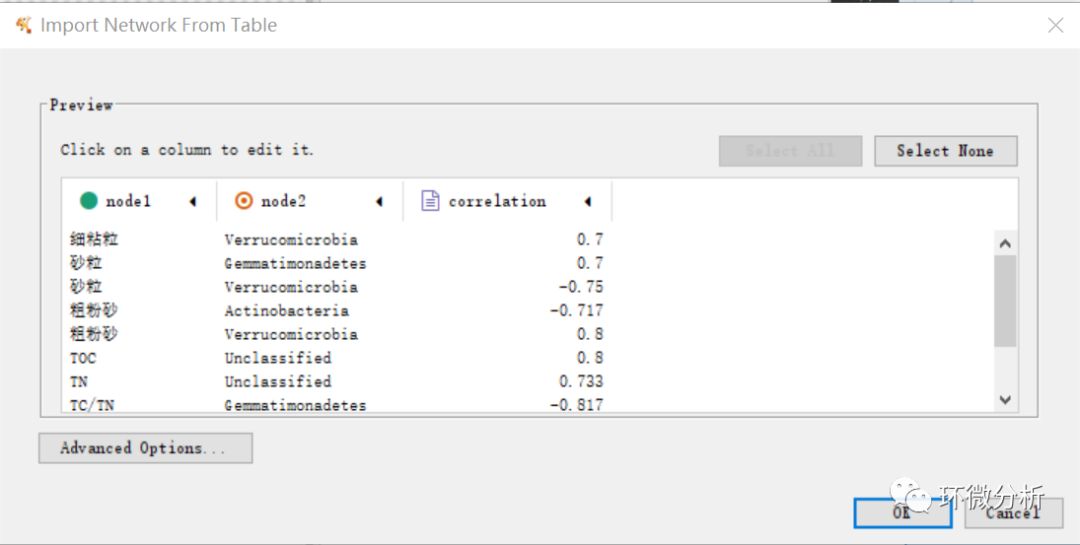
(3) 将node1(环境因子)设置为source node,将node2(细菌门水平注释结果)设置为target node,correlation(环境因子和微生物的sperman相关系数)设置为edge attribute,选择ok即完成数据导入;
(4) 得到初始网络图之后,点击左侧菜单栏的style对网络图进行美化;
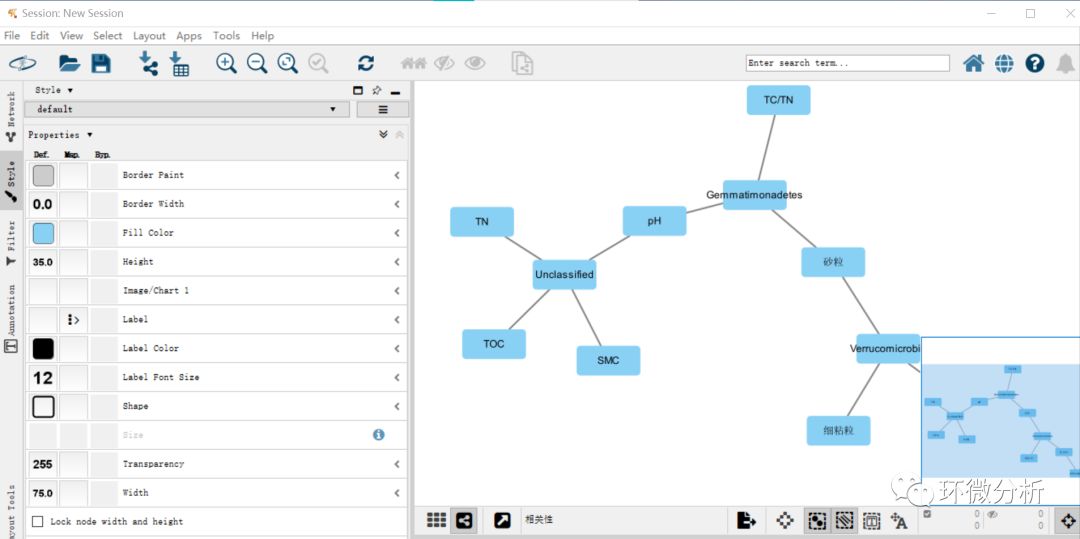
(5) 点击“Fill color”,根据name和discrete mapping对环境因子和微生物进行区分,完成后即可得到下图所示结果;
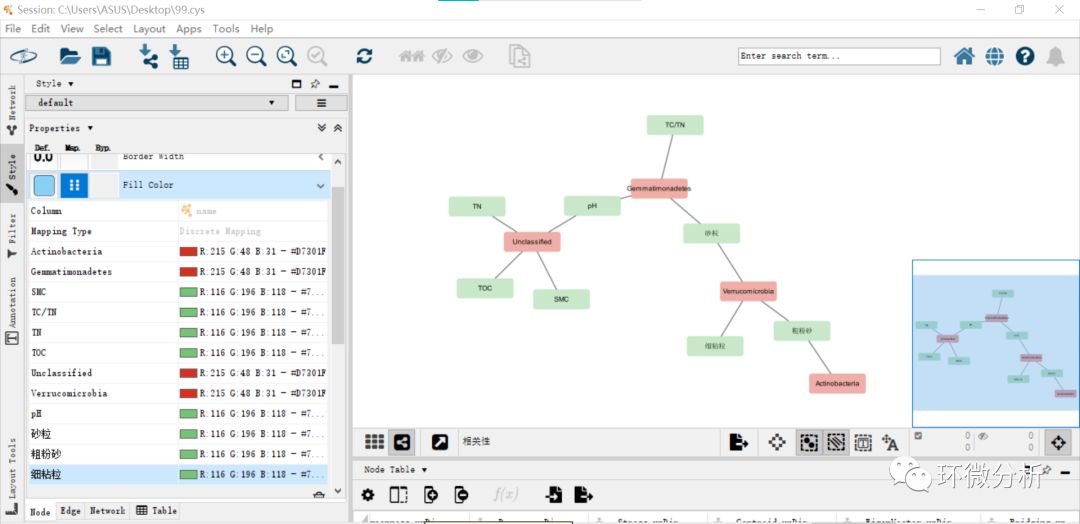
(6) 选择“shape”还可以设置不同node的形状,选择形状“Ellipse”。调整“Transparency”和“Width”可以调整node长宽比,勾选“lock node width and height”选项可以将node形状设置成正圆或正方形等。修改“Style”>“Node”>“Size”的值大小为35,并勾选“Lock node width and height”;
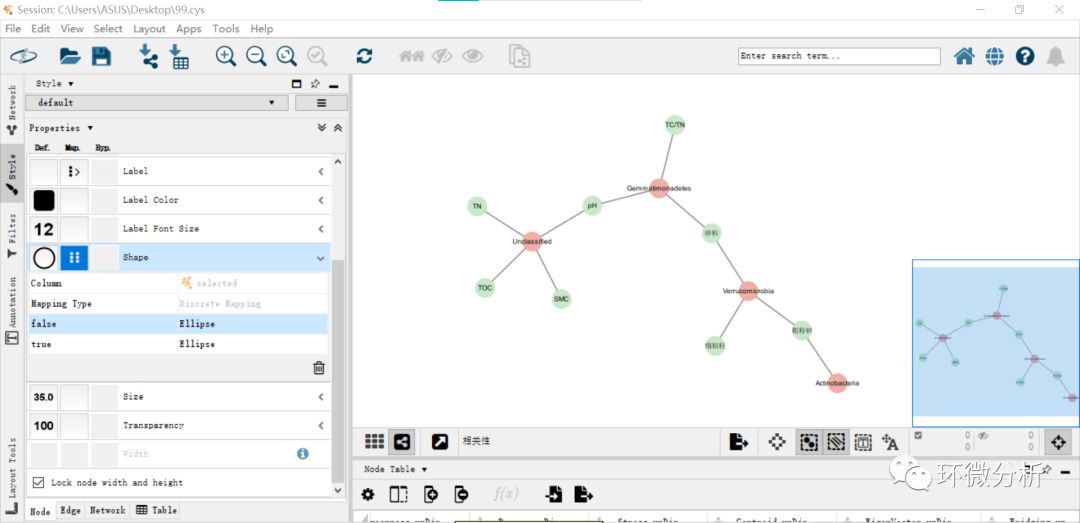
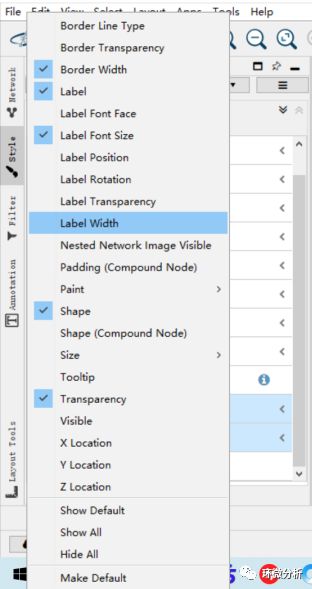
(7) 在style-node界面还可以设置很多参数,如字体、文字大小等。点击左上方的properties即可展示全部的可设置的参数,通过勾选来调用设置模块;
(8) 使用CentiScaPe 2.2插件计算每个点的权重(即连线数多少)。选择“Apps”>“App manager”进入应用管理界面,找到并选中CentiScaPe 2.2,点击“Install”进行安装;
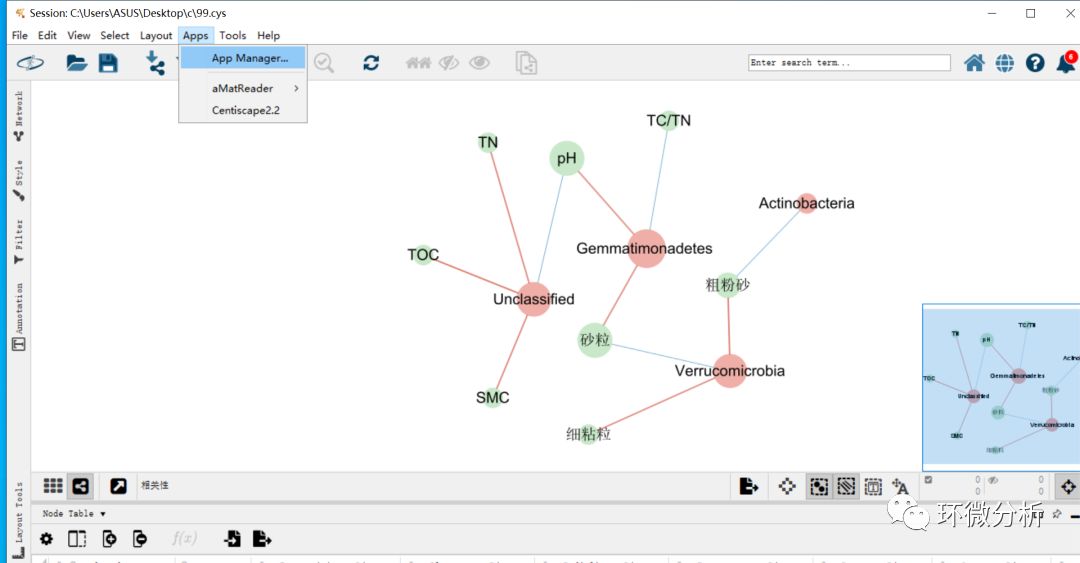
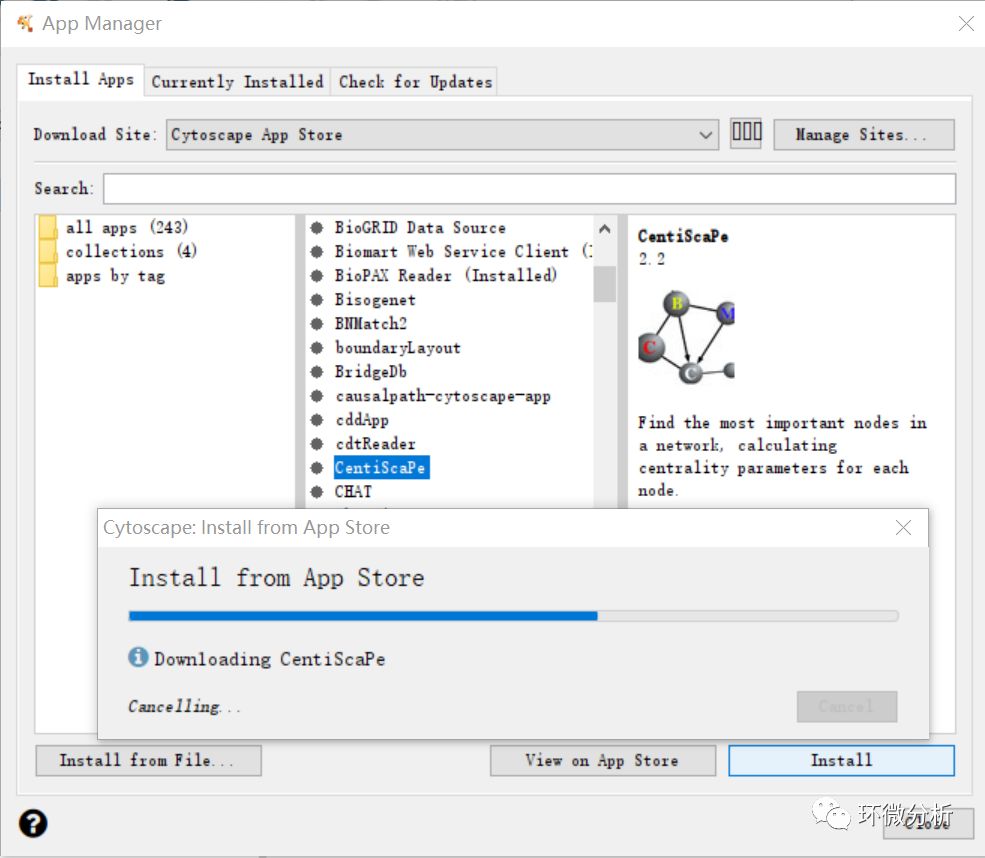
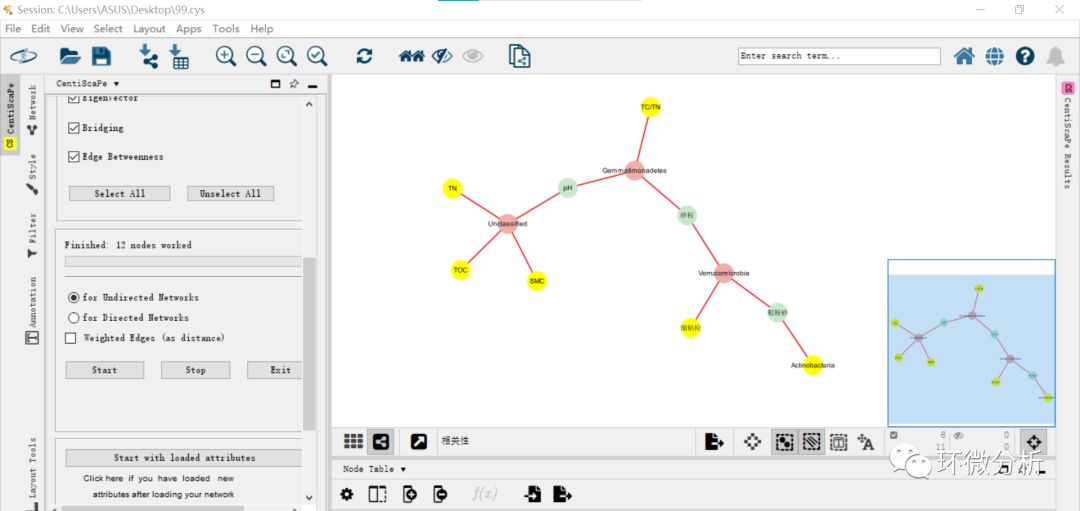
(9) CentiScaPe 2.2安装完成,按住control键,拖动鼠标全选显示框中所有noda。选择“Apps”>“CentiScaPe 2.2”。Implemented centralities框选择“Select All”,此外,选择“for Undirected Networks”。点击“Start”计算,如果是二次计算,会弹出提示框,点击“Start computation”继续即可;
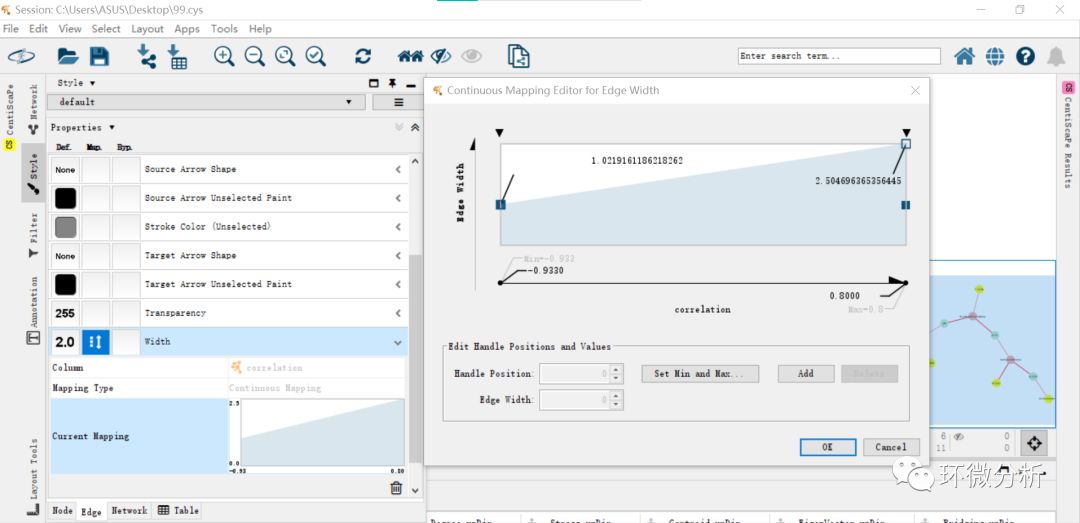
(10) 计算完权重后,点击“Style” >“Edge”>“Width”,Column设置成“correlation”,即可让连线的大小随权重而变化。双击“Current Mapping”可以对线条粗细分布进行调整;
(11) 点击stroke color(unselected)可以将节点之间的连线设置成不同颜色,即将负相关关系设置成蓝色,将正相关设置成红色,便于区分;
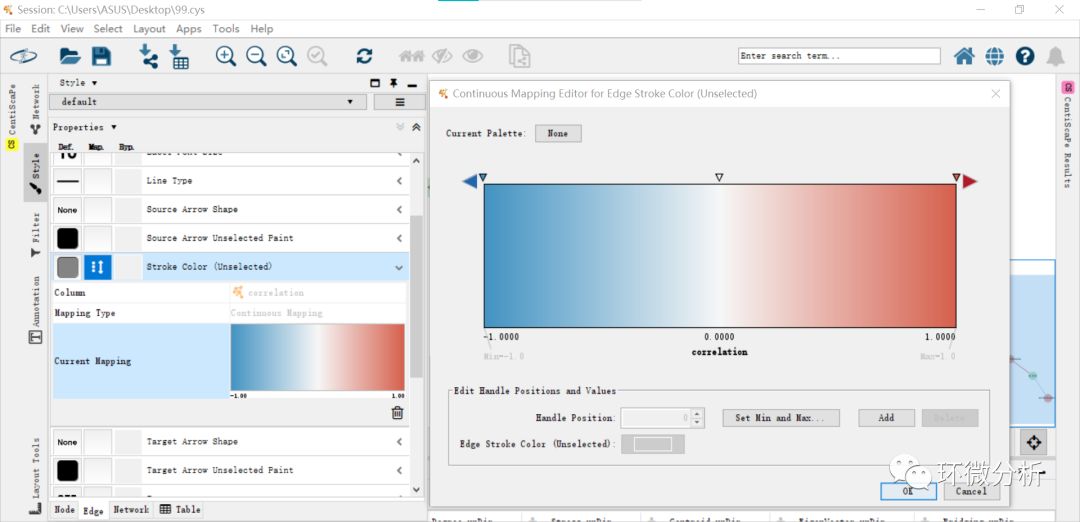
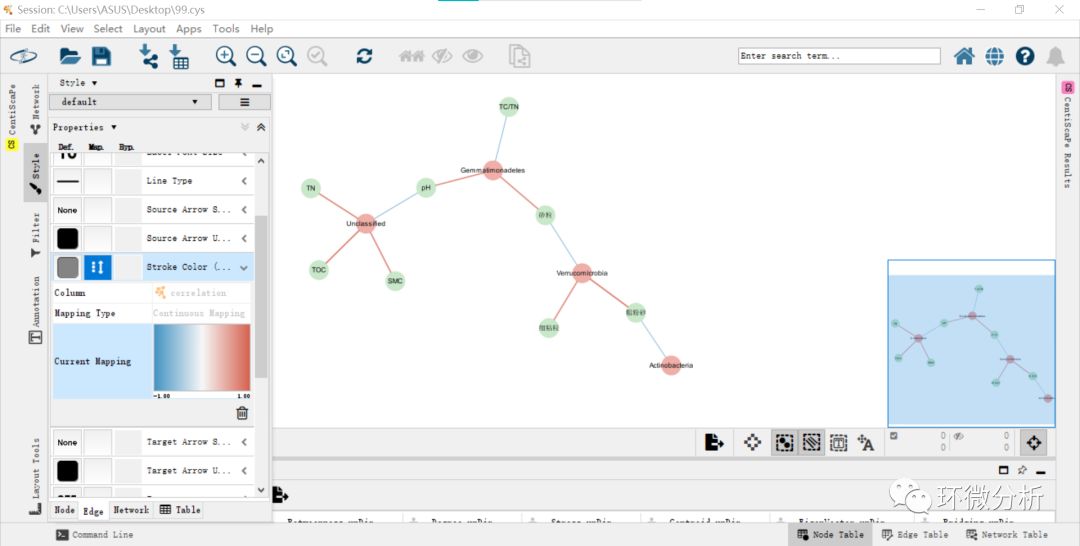
(12) 图形的设置完成后,也选择“Layout”>“…”来选择合适的分布类型。点击不同的设置,即可出现不同的分布。如果想要更多的分布方式,则可安装yfiles layout algorithms插件。点击apps>app manger安装。此外,还可以在style>Node调整标签尺寸以及根据CentiScaPe 2.2的计算结果(度)来调整Node的大小。完成后,即可得到以下图形;
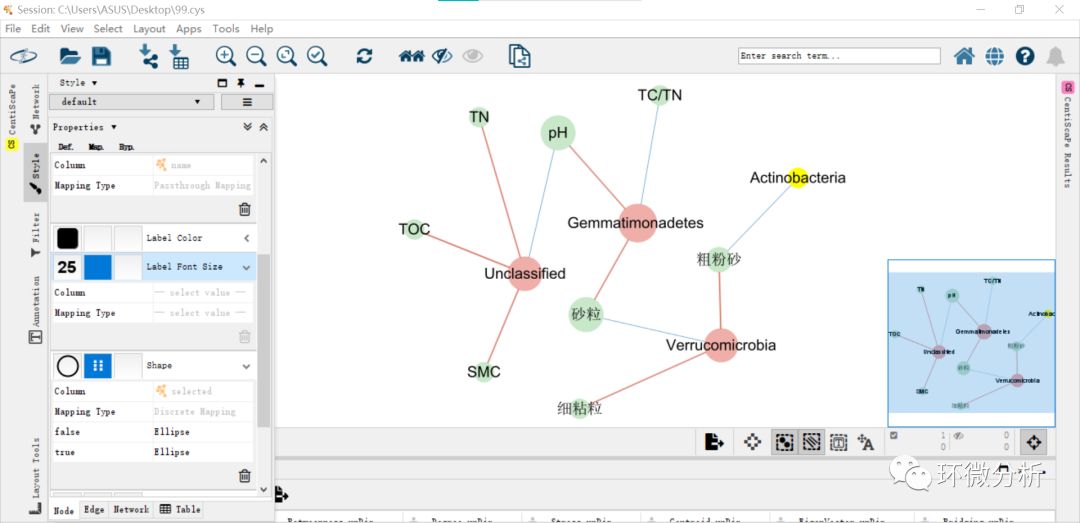
(13) 在导出图形之前,点击“Fit Content”以保证所有数据都在视图之内,点击“file”>“export”导出图片;
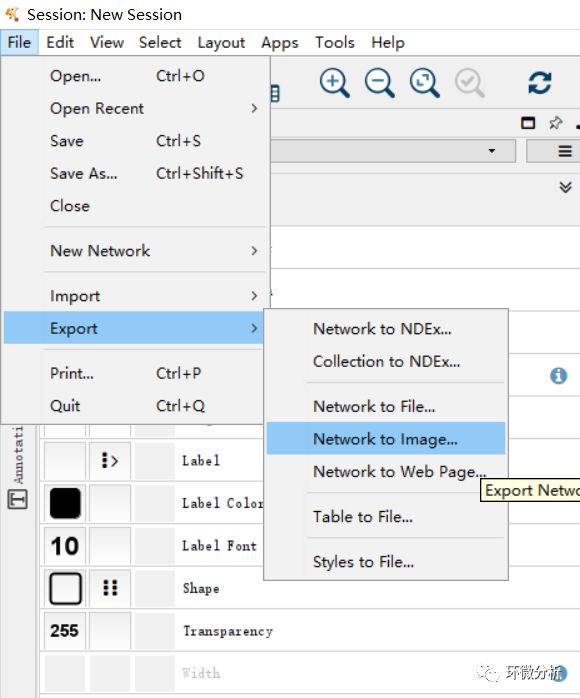
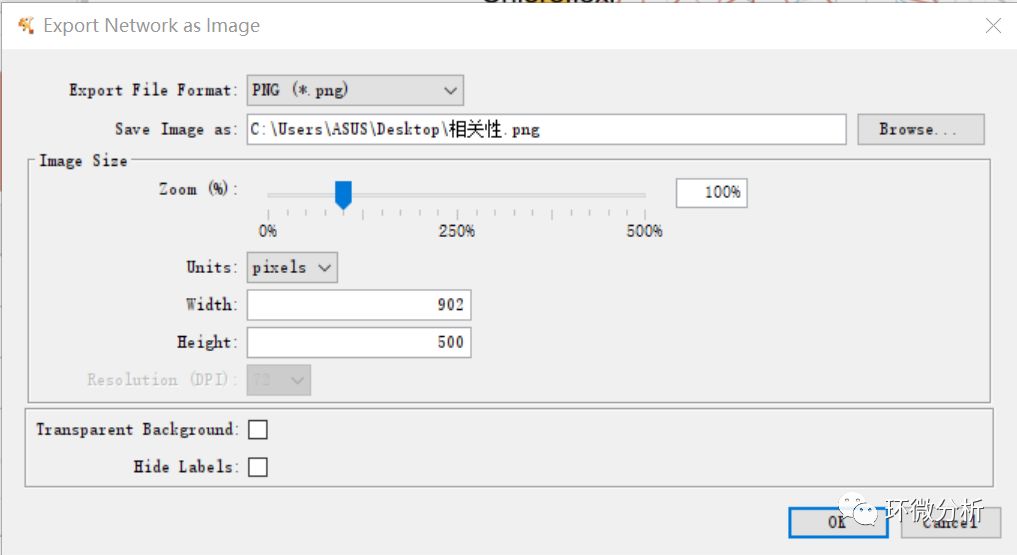
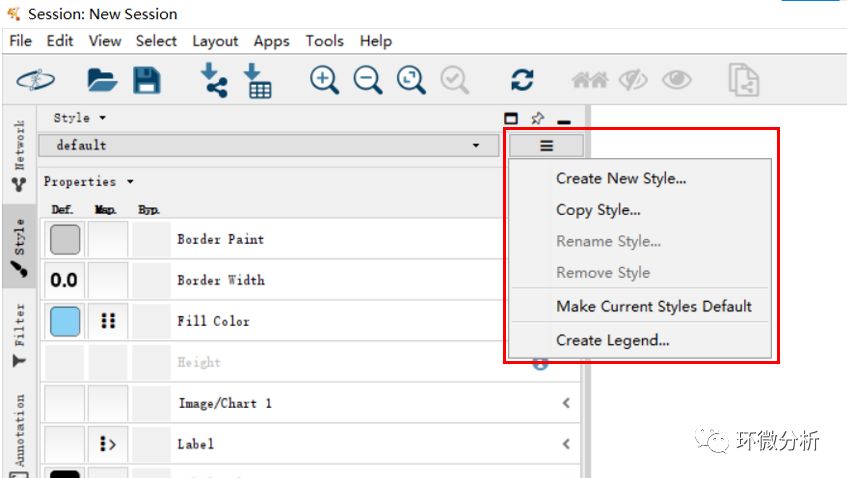
(14) 点击“Options...”>“Create Legend…”,导出图例信息;
(15) 根据自己的需要使用AI软件修改调整图片,结果如下图所示:
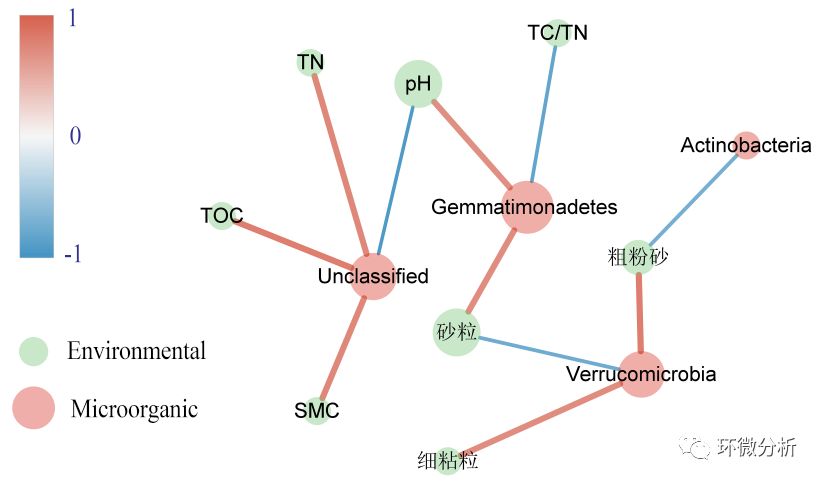
图表解读:图中绿色节点代表环境因子,红色节点代表物种数据,节点大小代表度的大小,即是连线多少。连线代表具有相关性,蓝色代表负相关,红色代表正相关,连线颜色深浅与线条粗细表明相关性大小,颜色越深,相关性越强;线条越粗,相关性越强。从图中可以看出,Unclassified与TN、TOC、SMC呈现正相关,与pH呈现负相关;Gemmatimonadetes门与砂粒、pH呈现正相关,与TC/TN呈现负相关;Verrucomicrobia门与细粘粒、粗粉砂呈现正相关,与砂粒呈现负相关;Actinobacteria与粗粉砂呈现负相关。
———— Gephi
一、软件安装
(1) 登录官网https://gephi.org/,点击“Download FREE”下载安装Gephi;
(2) 下载完成后,双击执行文件gephi-0.9.2-windows.exe进入安装向导窗口,点击“Next”继续;
(3) 翻看协议,勾选“I accept the agreement”,点击“Next”;
(4) 选择合适的安装位置,点击“Next”;
(5) 选择Mebu 文件夹,保持默认,点击“Next”;
(6) 选择需要添加的文件,保持默认,点击“Next”;
(7) 确认安装信息,点击“Install”开始安装;
(8) 安装成功,点击“Finish”退出安装;
二、数据处理
(1) 准备点数据文件
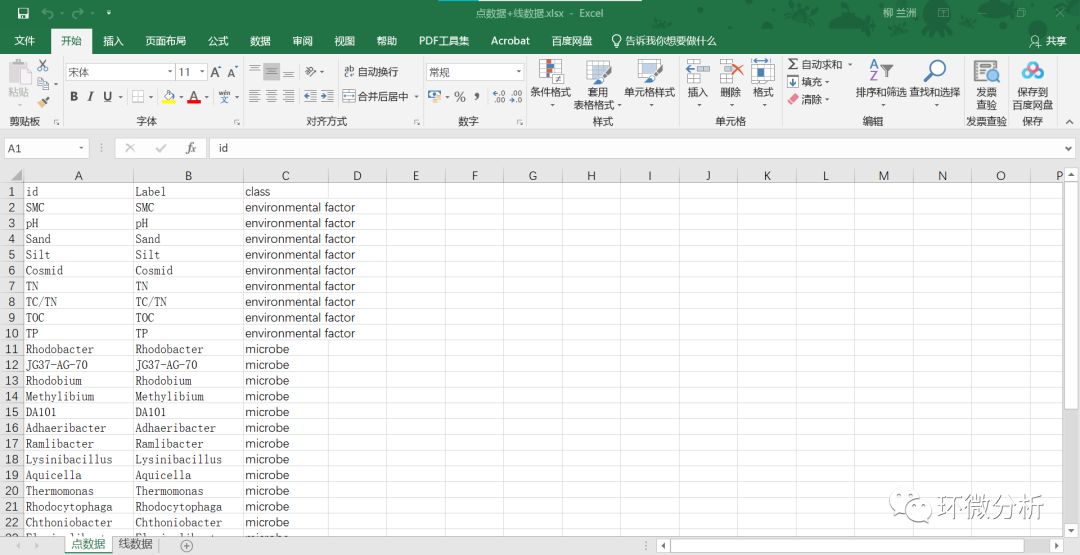
先对数据正态性检验,存在非正态分布的微生物数据因子。对理化数据以及微生物属水平物种丰度数据进行斯皮尔曼相关性分析,挑出所有相关性显著的数据对,做成点数据与线数据文件。“点数据”工作表A列必须是“id”为列头,是每一个用于构建网络结构节点的唯一标识;后面的列是根据自己的需要添加的,示例数据添加了标签列Label(点数据标签,与id列保持一致)和分类信息列class(用于区分微生物物种丰度数据与理化因子数据)。编辑好的数据文件保存为.xlsx形式,分批次导入点数据与线(边)数据;也可以另存为.csv文件,方便一次性导入;
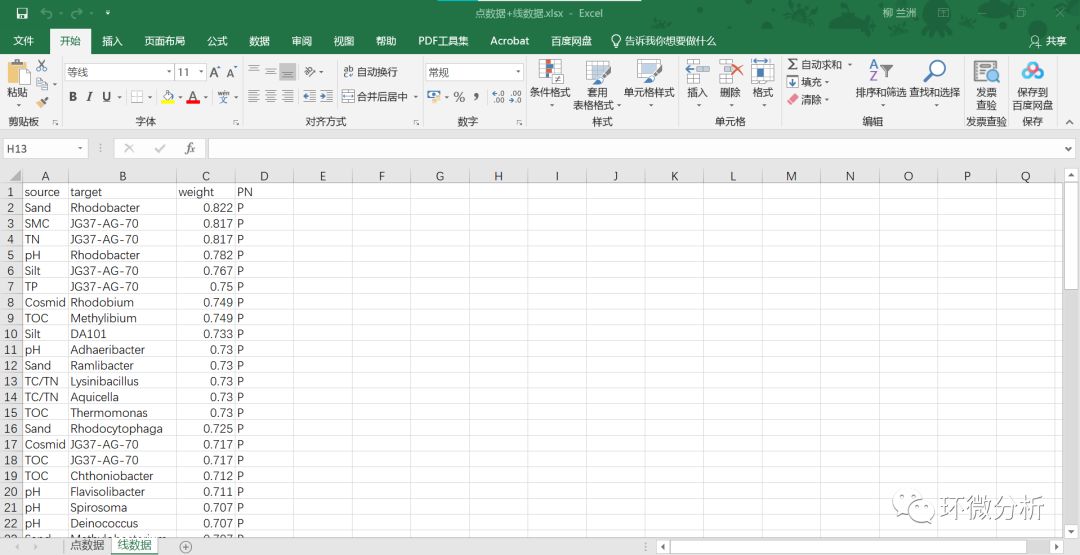
(2) 准备线(边)数据文件 “线数据”工作表A列与B列必须分别以“source”与“target”为列头,其中第一列是理化数据指标,第二列是与第一列理化指标有显著相关性的微生物物种数据;C列以“weight” 为列头,“weight”列填充的数据是前两列指标(研究对象)对应的相关性系数;后面的列不是必须的,第四列 “PN”列是代表的是相关性正负符号,其中P代表正号(positive),N代表负号(negative);
三、作图流程 (1) 打开Gephi,选择“文件”>“打开”,弹出文件导入对话框,选择文件“点数据+线数据.xlsx”,点击“打开”;
(2) 弹出导入向导对话框,先导入的是点数据,表单选择“点数据”,导入数据选择“节点表格”,查看“预览”框确定导入是否正常,点击“下一步”;
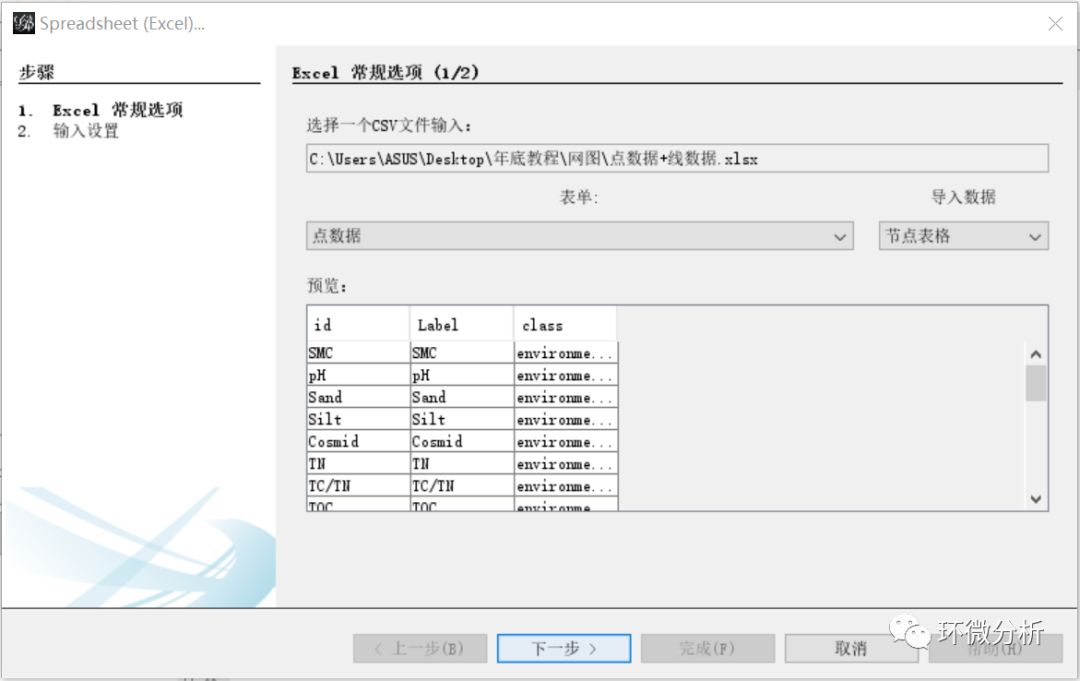
(3) 在“输入设置”界面,保持默认,点击“完成”继续;
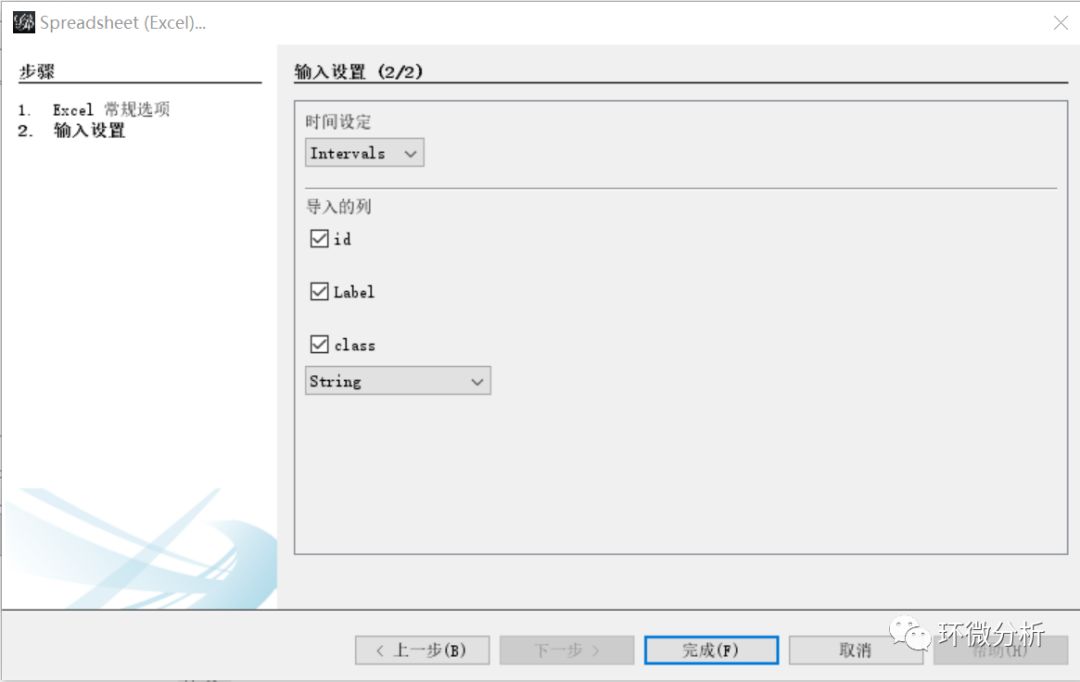
(4) 查看“输入报告”,图的类型选择“混合的”,选择“New workspace”,点击“确定”完成点数据导入;
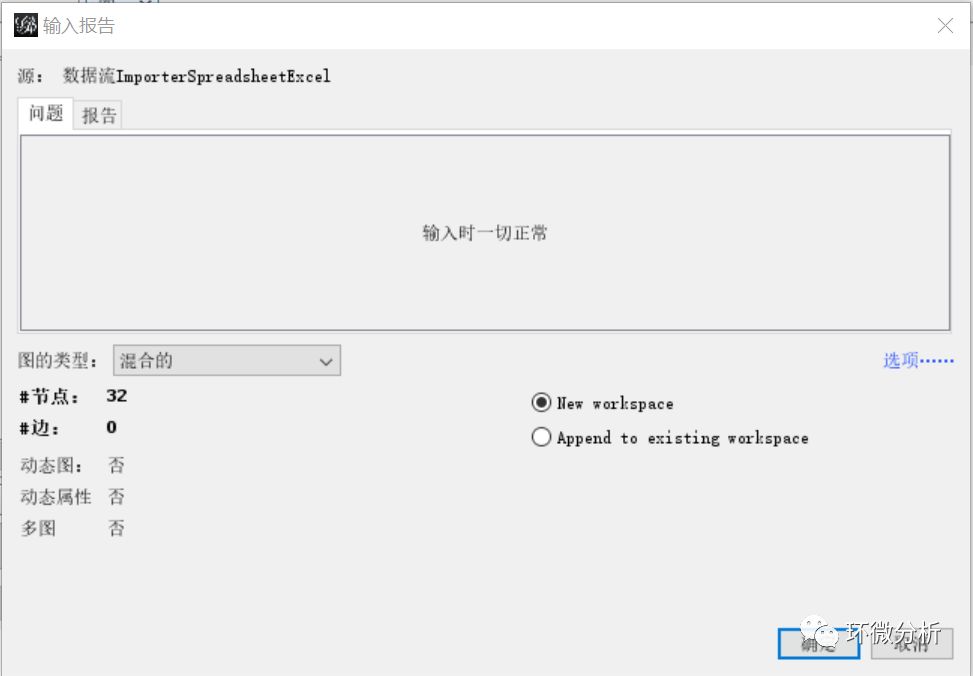
(5) 再次在主界面选择“文件”>“打开”,弹出文件导入对话框,选择文件“点数据+线数据.xlsx”,点击“打开”即弹出导入向导对话框,现在导入的是线数据,表单选择“线数据”,导入数据选择“边表格”,查看“预览”框确定导入是否正常,点击“下一步”;
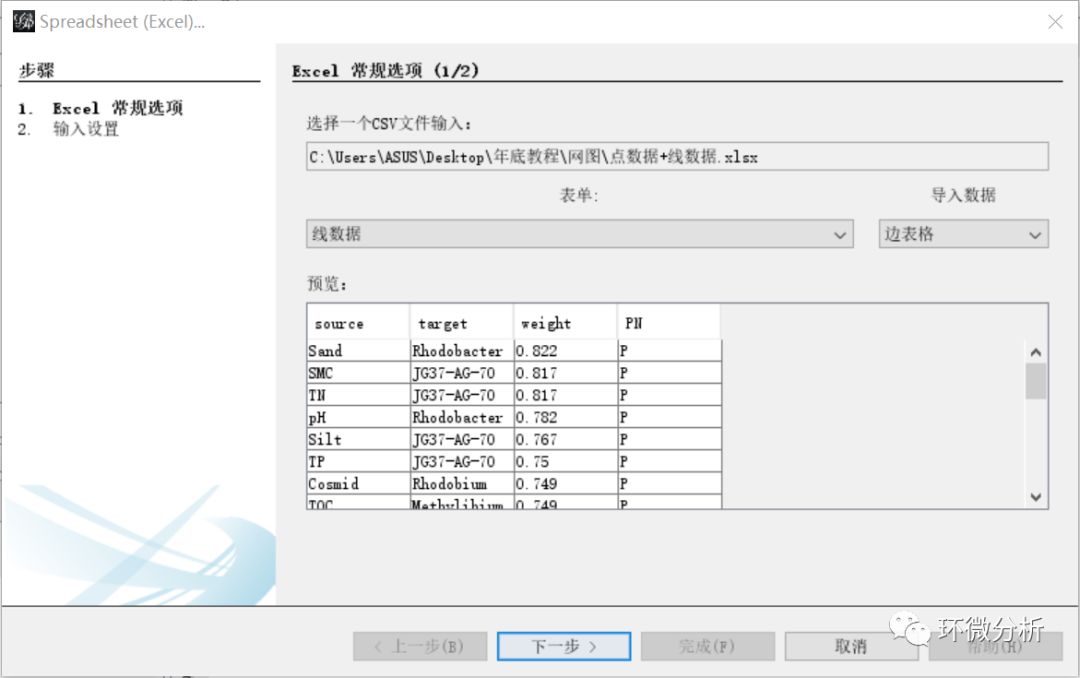
(6) “输入设置”界面,weight选择“Float”格式,其他保持默认,点击“完成”继续;
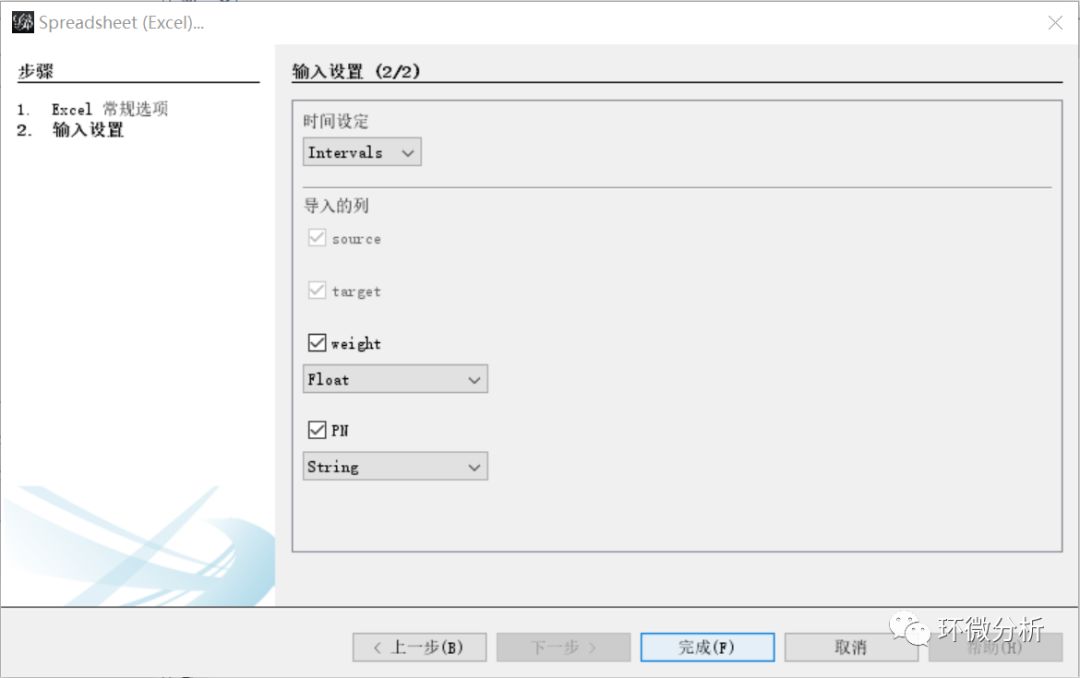
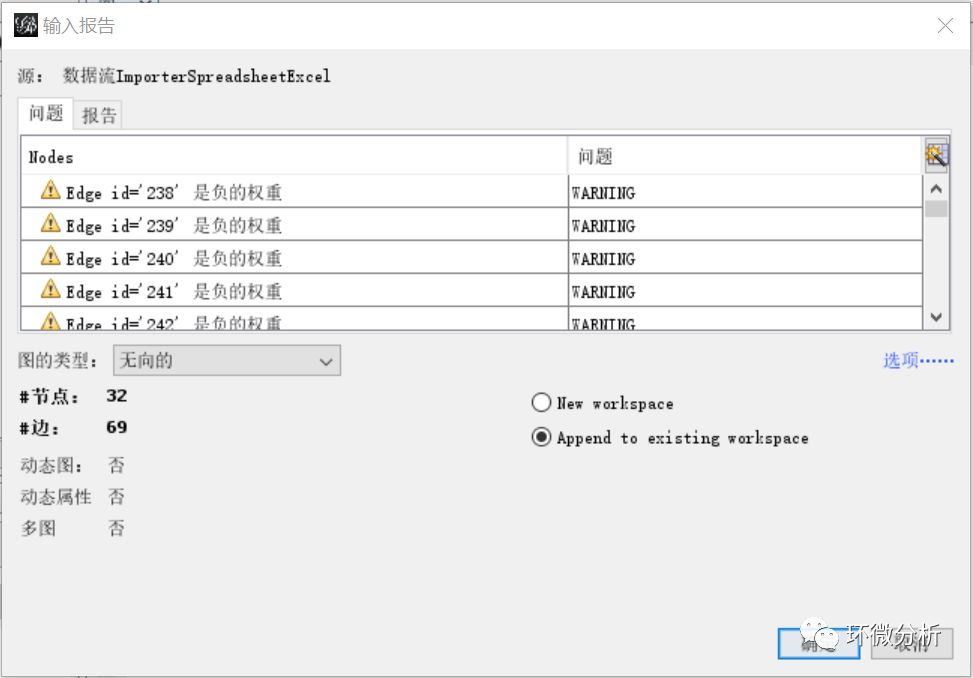
(7) 查看“输入报告”,这里提示了有“负的权重”问题,这是因为相关系数存在负数,可以忽略。图的类型选择“无向的”,选择“Append to existing workspace”,点击“确定”完成线数据导入;
(8) 导入点数据与线数据后的初始图形如下:
(9) 选择“概览”> “外观”>“布局”>“Fruchterman Reingold”,点击“应用”将微生物物种数据与理化指标布局成圆环状,布局成功后需要点击“停止”结束计算;
(10) 选择“概览”>“外观”>“节点”>“颜色”> “Partition”>“class”,点击“应用”即使用不同颜色将微生物物种指标与理化指标区分开;
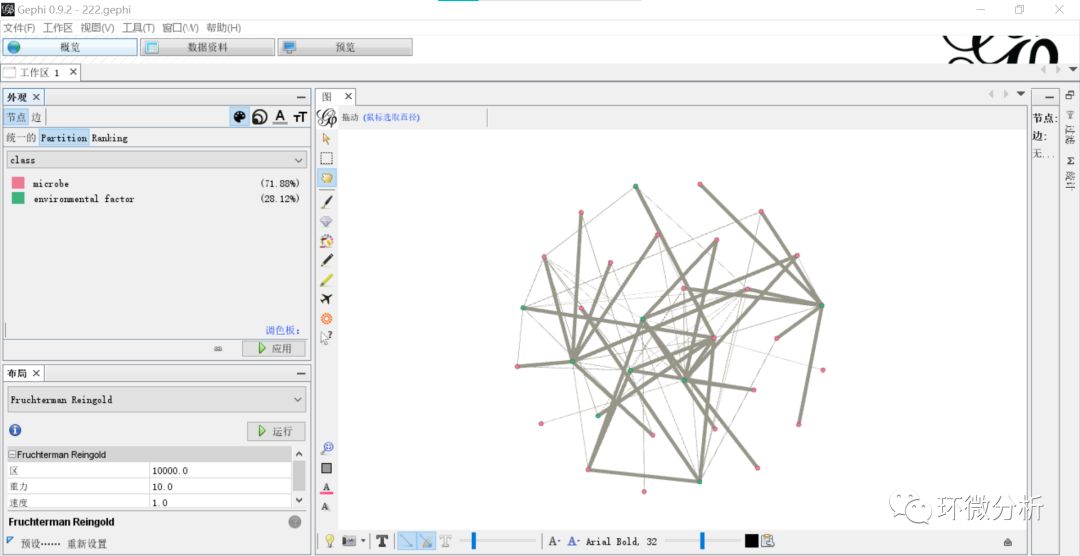
(11) 选择“概览”>“外观”>“节点”>“大小”>“Ranking”>“度”,修改最小尺寸与最大尺寸,这里分别是30,70,点击“应用”即可将节点大小与度的大小关联起来;
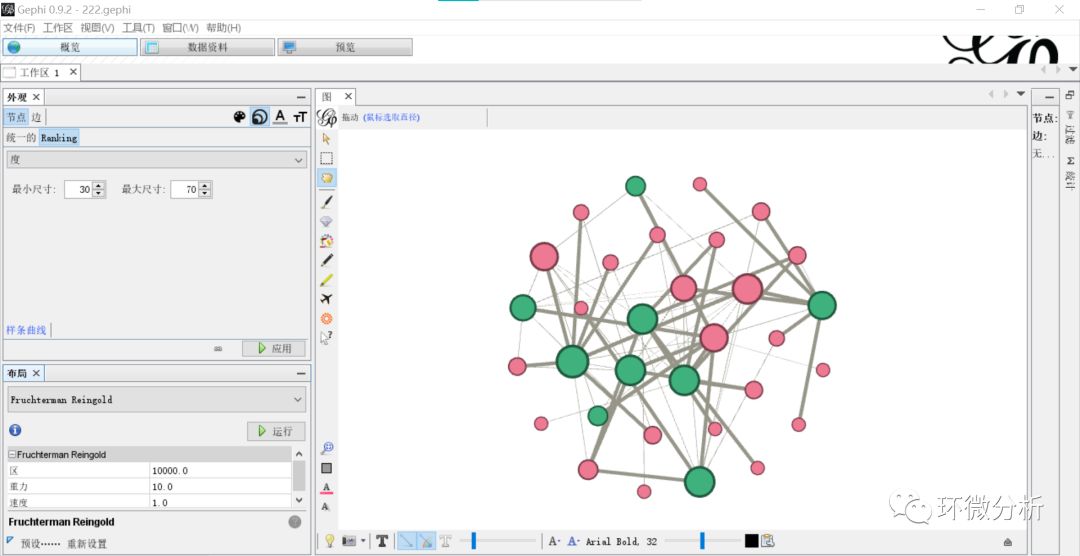
(12) 修改“概览”>“外观”>“边”>“Partition”为“pn”,即符号正负;
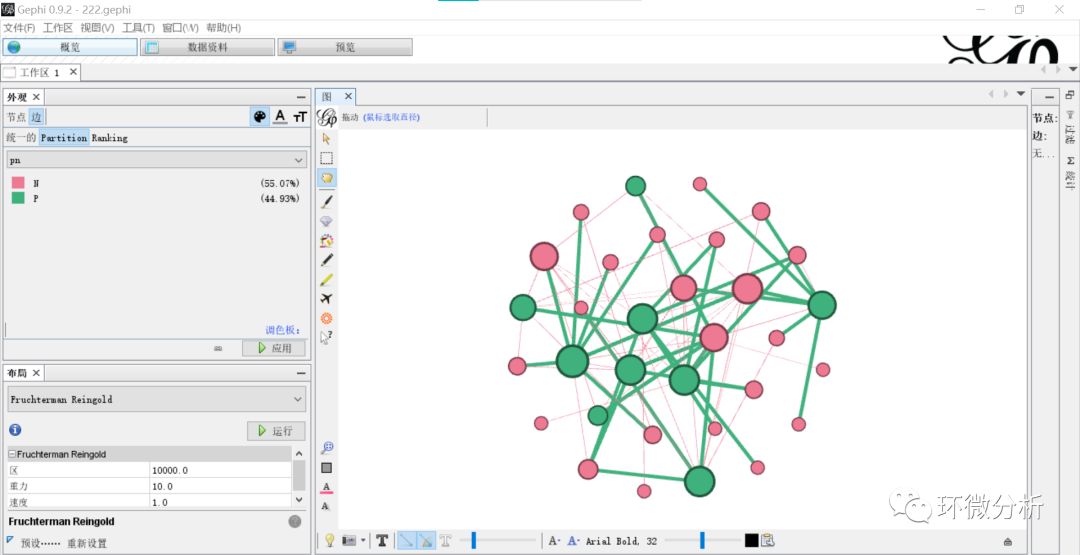
(13) 选择“窗口”>“统计”,界面右侧出现参数统计工作框,依次统计所有参数统计量,出现对话框,出现“连通分量设置”提示框均选择“无向”,点击“确认”计算,计算结束关闭对话框即可;
提示:如果点击“显示标签”,Label标签未出现。可以查看“数据资料”界面,如果原因是见第二列“Label” 列无数据;选择页面下方“复制数据到其他列”>“label”(标签列),即选中需要被复制的列;在弹出对话框选择Label列,点击 “好”完成复制;查看“数据资料”页面,可见Label列已经填充了原来的lable列数据了。如果在编写点数文件时,lable列的列头改写成“Label”,就可以轻松避免这个问题;
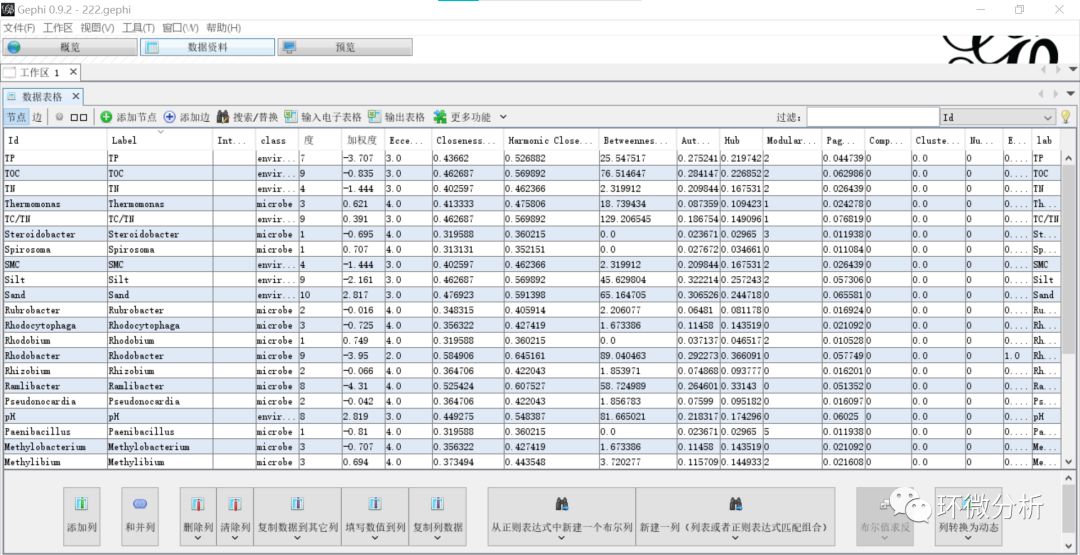
(14) 在“预览”界面,修改“设置”>“节点”>“边框宽度”为0,勾选“设置”>“节点标签”>“显示标签”,不勾选“设置”>“节点标签”>“比例大小”,调整“设置”>“节点标签”>“字体”格式,点击“刷新”出现标签;
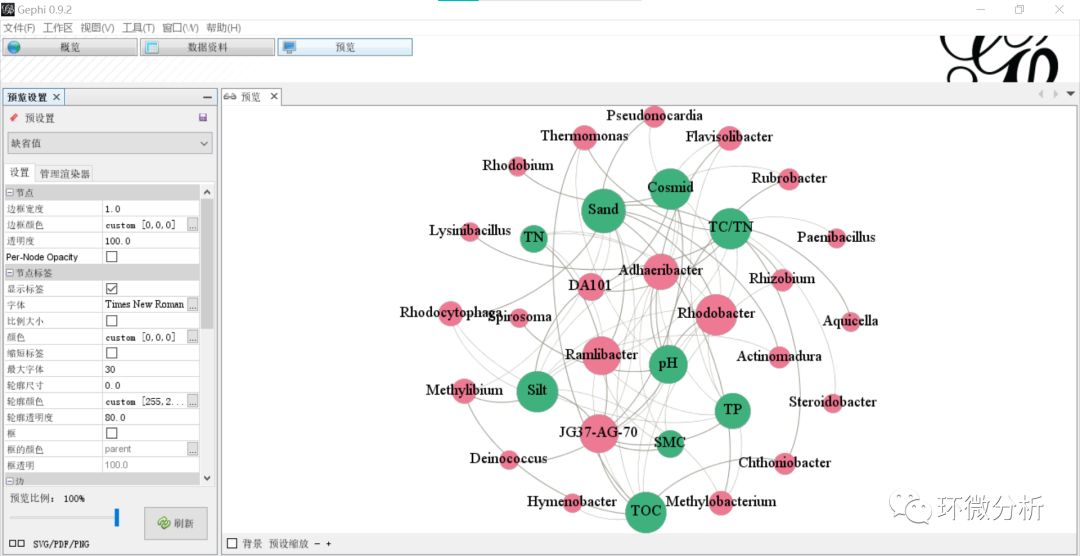
(15) 选择“预览”>“设置”>“厚度”修改为3.0,“颜色”选择“原始的”,点击“确认”;
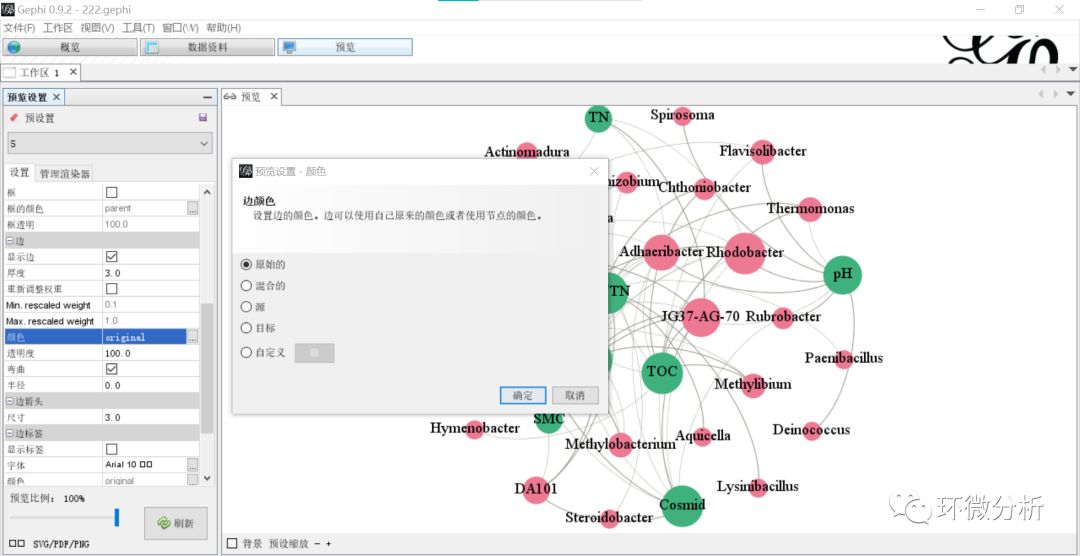
(16) 再点击“刷新”进行设置更新,结果如下:
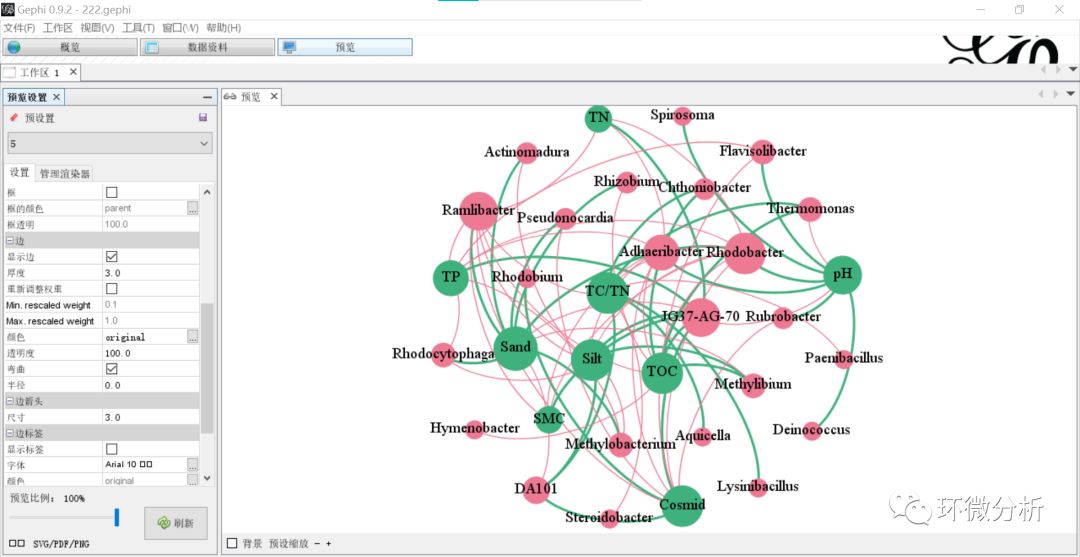
(17) 也可以设置成根据关系源赋色。进入“数据资料”界面,选择“添加列”,弹出设置框,标题输入“lab”,点击“好”完成添加;
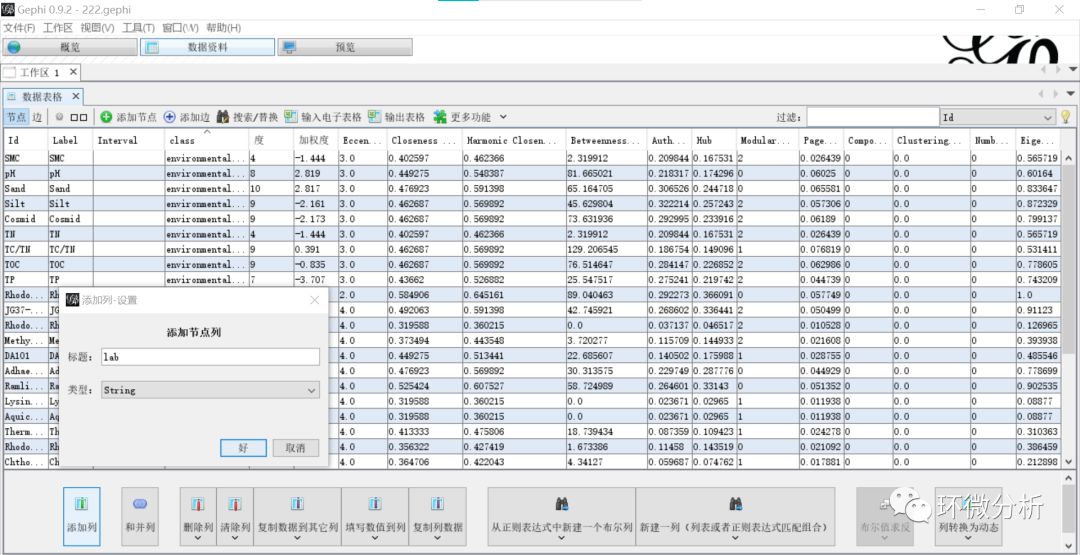
(18) 选择“复制数据到其它列”>“Label”,弹出对话框;
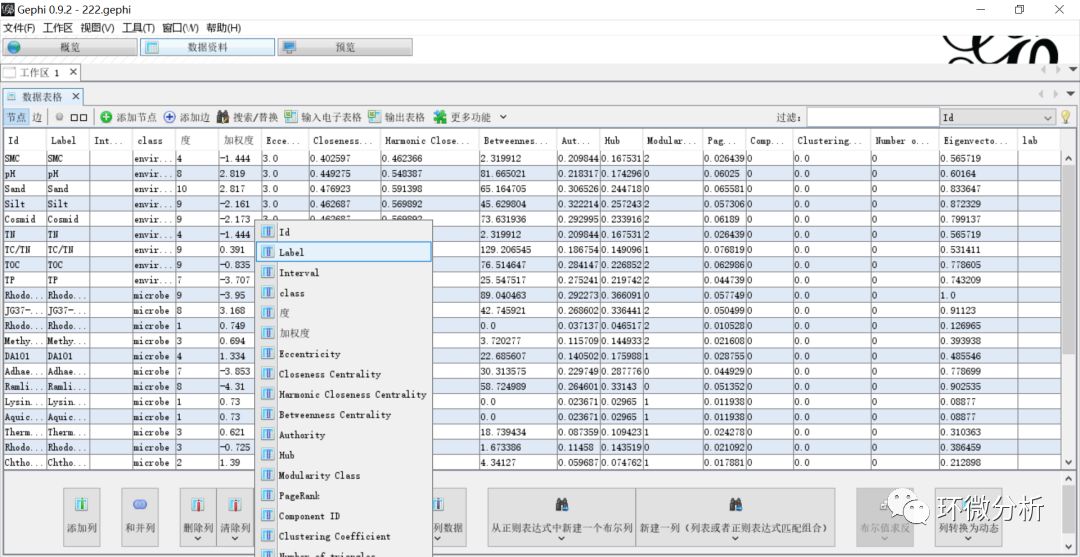
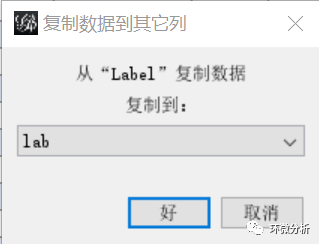
(19) 在“复制数据到其它列”对话框,输入“lab”,点击“好”完成复制;
(20) 查看工作区的数据,可见已经成功添加了“lab”列与拷贝了“Label”列数据;
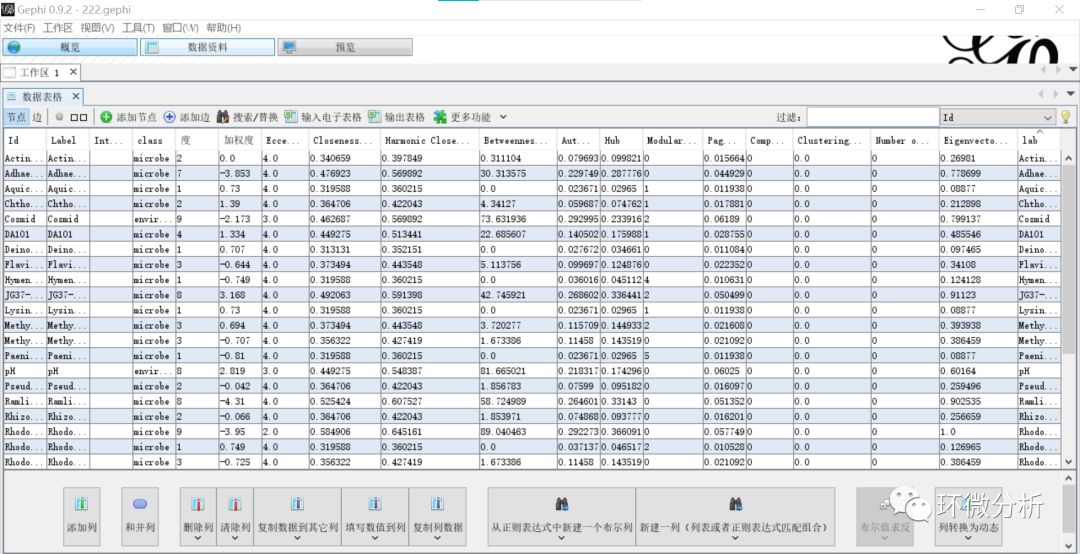
(21) 选择“概览”>“外观”>“节点”>“Partition”>“lab”,即将节点颜色设置成随着节点“lab”列数据改变;
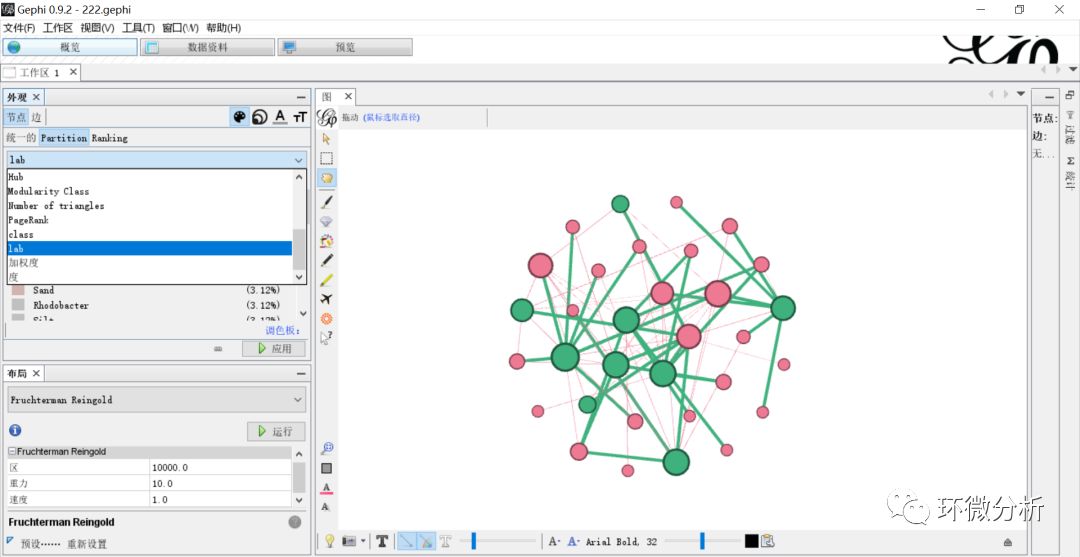
(22) 点击“概览”>“外观”>“节点”>“Partition”> “调色板”>“生成”;
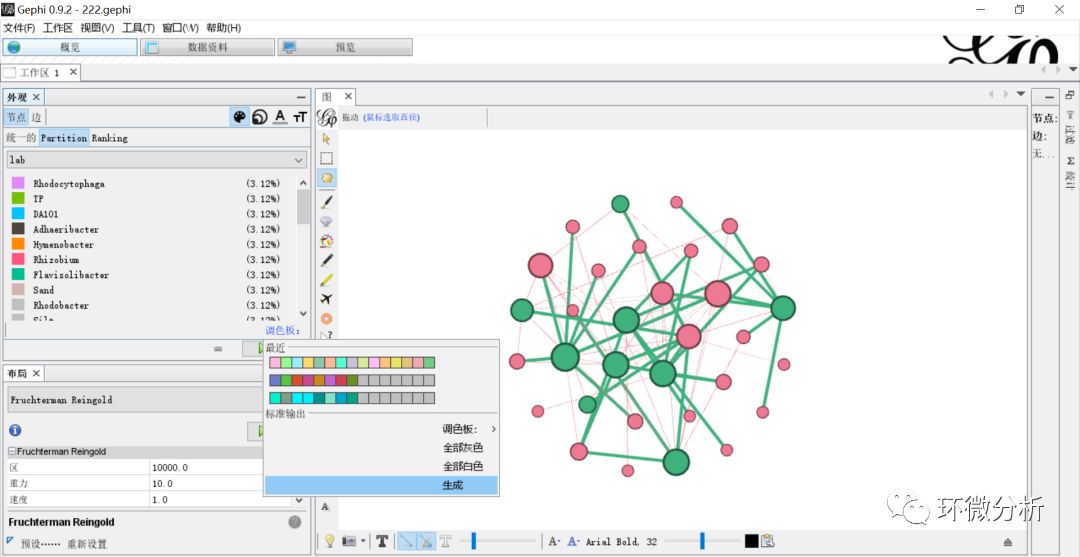
(23) 输入复制的数量“32”,这个可以根据你的Id数目进行设置分配,Id数目即是网图中所有节点的数目;
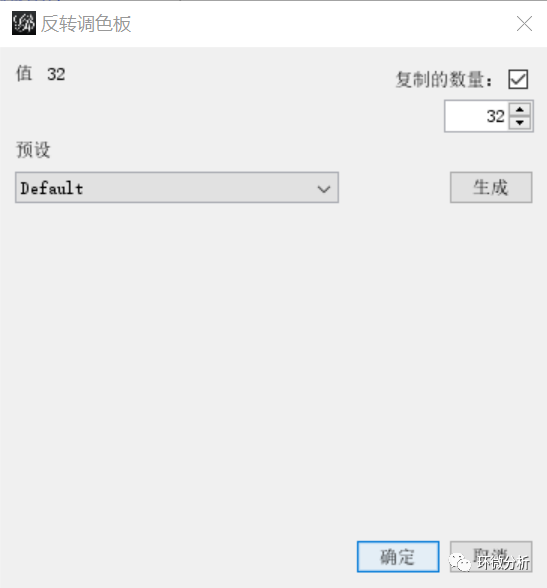
(24) 生成调色板后,选择生成的目标调色板,点击“应用”;
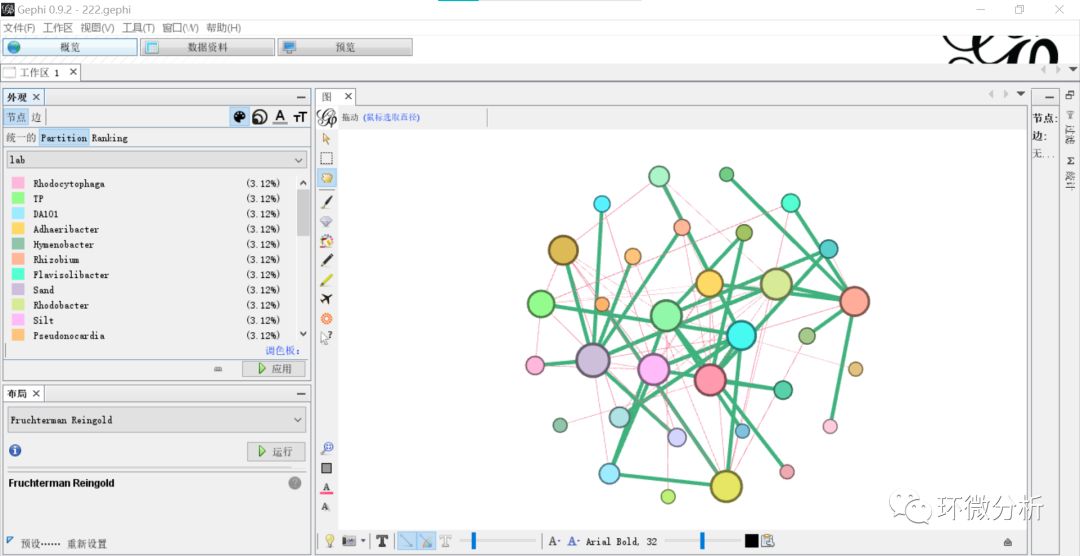
(25) 返回“预览”界面,选择“设置”>“边”>“颜色”>“源”,点击 “确认”;
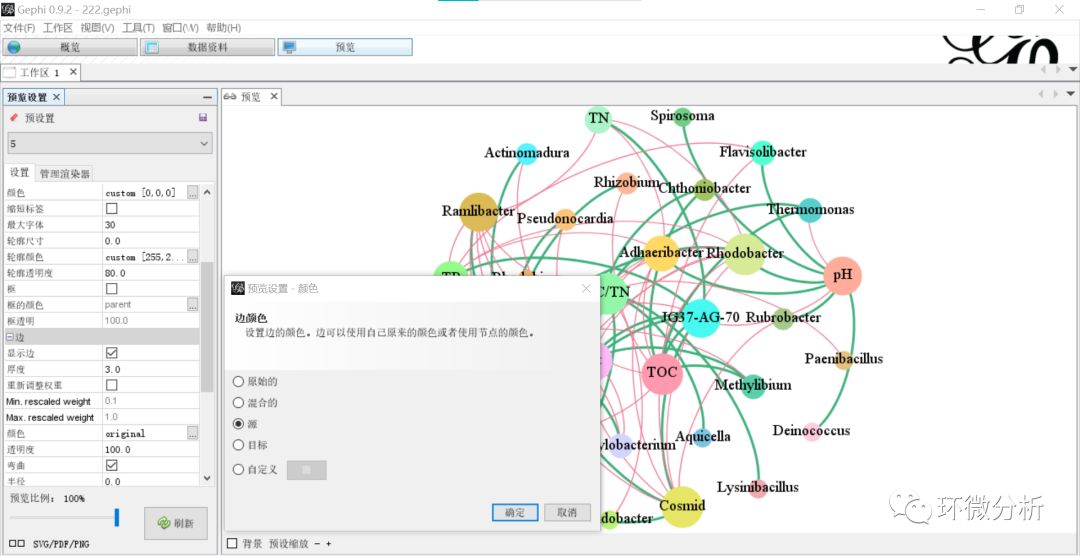
(26) 点击“刷新”,输出结果如下:
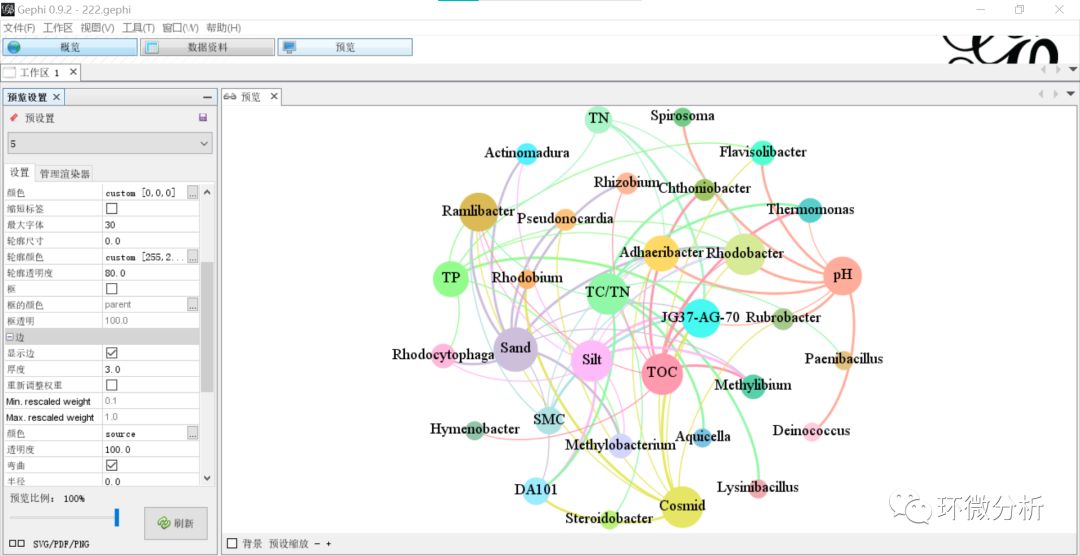
(27) 点击界面左下方“SVG/PDF/PNG”按钮,弹出文件输出框,选择合适的图片格式进行输出;
(28) 导出图片,使用AI软件修改后的效果图如下。
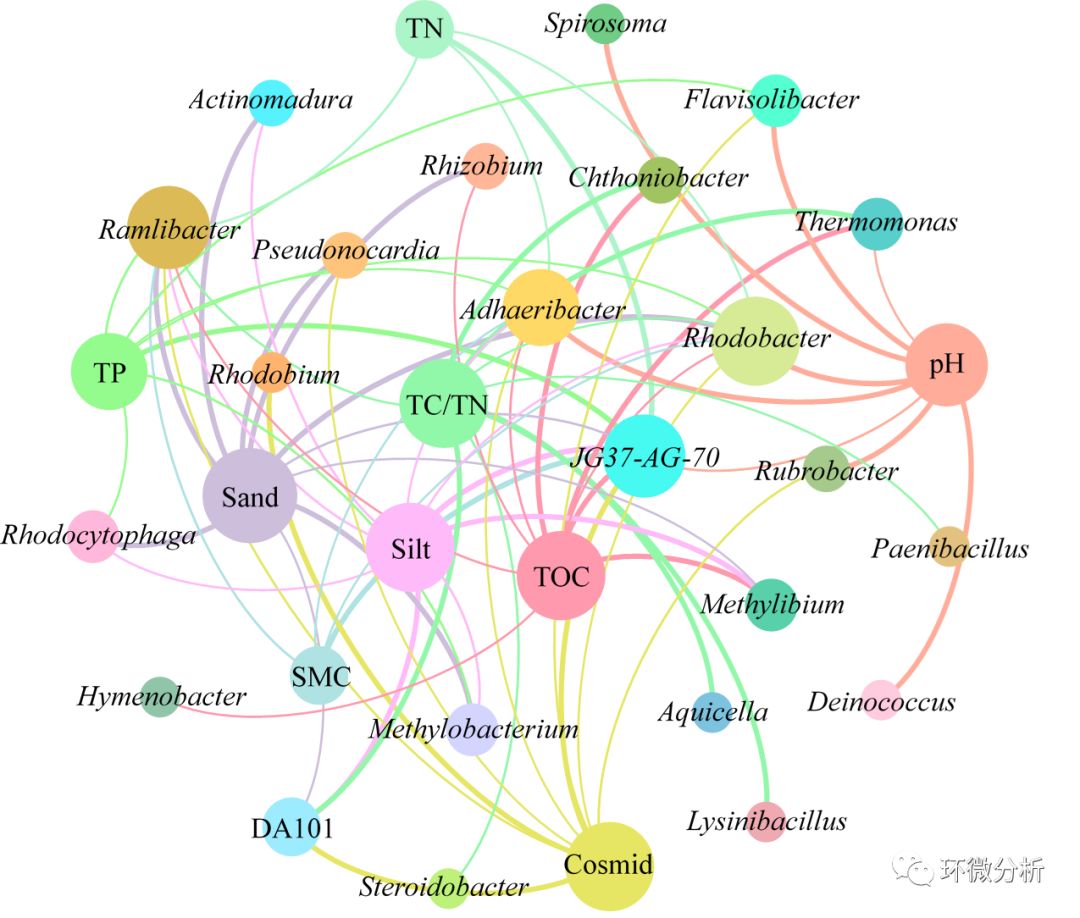
图形解读:圆圈的大小代表相关的连线的数目;线条的粗细表示相关性大小,相关系数的绝对值越大,线条就越粗。由图可见,TC/TN、Sand、TOC、pH和Cosmid5个环境变量与微生物种属关联度较大,Ramlibacter属、Adhaeribacter属与JG37-AG-70属与环境因子关联度较大;其中,环境因子pH与微生物种属的Rhodobacter、Adhaeribacter、Flavisolibacter、Spirosoma、Deinococcus和Rubrobacter关联性较强;环境因子Sand与微生物的Rhodobacter、Ramlibacter、Rhodocytophaga、Methylobacterium、Pseudonocardia、Actinomadura、Rhizobium、DA101关联性较强。
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!

















 393
393

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









