







本文主要介绍生物信息学软件 Mothur基于OTU的分析流程。
数据分析
接下来实际分析数据。关注基于OTU的数据集,与使用ASV或phylotypes进行的分析基本相同。另外,请记住,当比较早期和晚期样本时,最初的问题与这些样本的稳定性和群落结构的变化有关。请记住,组名要么是F或M(动物性别),然后是数字(动物数量),最后是D和三位数(断奶后天数)。为了简单起见,重命名count、tree、shared和consensus taxonomy文件。
mothur>rename.file(taxonomy=stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.0.03.cons.taxonomy,shared=stability.trim.contigs.good.unique.good.filter.unique.precluster.pick.pick.pick.opti_mcc.shared)

现在要做的是查看每个样本中有多少个序列。使用count.groups命令执行此操作:
mothur>count.groups(shared=stability.opti_mcc.shared)
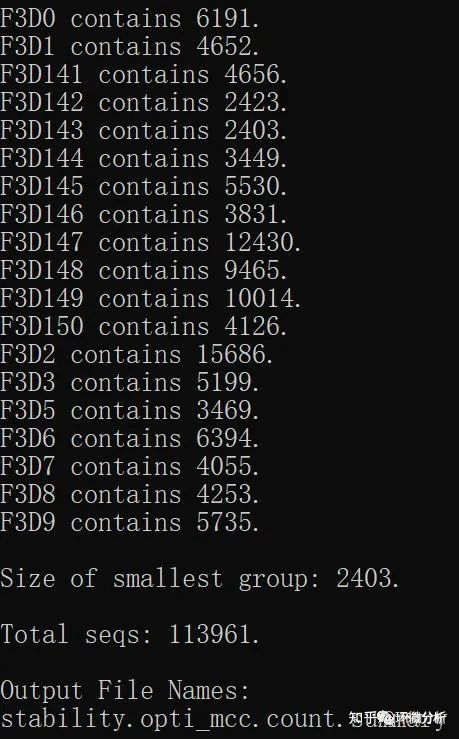
可以看到最小的样本中包含2403条序列,这是一个合理的数字。无论如何,对数据进行二次抽样和稀疏是一件重要的事情。使用sub.sample命令生成一个用于分析的二次抽样文件:
mothur>sub.sample(shared=stability.opti_mcc.shared, size=2403)

基于OTU的分析
Alpha多样性
通过分析样本的alpha多样性开始数据的实际分析。首先生成稀疏曲线,描述观察到的OTUs数量与采样工作量的关系。使用rarefaction.single命令执行此操作:
mothur>rarefaction.single(shared=stability.opti_mcc.shared, calc=sobs, freq=100)

这将生成以*.rarefaction结尾的文件,该文件可以在图形软件包中进行绘制。稀疏(rarefaction)并不是衡量丰富程度的标准,而是衡量多样性的标准。如果考虑两个群落的丰富度相同,但均匀度不同,那么在对大量个体进行采样后,它们的稀疏曲线将逐渐接近相同的值。由于它们具有不同的均匀度,因此曲线的形状将不同。因此,选择一些个体来截断稀疏曲线并不能使研究人员基于丰富度来比较样本,而是基于样本的多样性。最后,使用summary.single命令得到一个包含序列数(number of sequences)、样本覆盖率(Coverage)、观测OTU数(observed OTUs)和逆Simpson多样性(Inverse Simpson diversity)估计的表。为了使所有内容标准化,从每个样本中随机选择2403个序列1000次并计算平均值(注意:如果设置subsample=T,则它将使用最小库):
mothur>summary.single(shared=stability.opti_mcc.shared,calc=nseqs-coverage-sobs-invsimpson,subsample=T)

这些数据被输出到一个名为“stability.opti_mcc.groups.ave-std.summary”的文件中的表中。打开文件可以看到样本覆盖率均高于97%,表明对群落进行了很好的抽样。绘制样品的丰富度或多样性将表明不同动物之间或早期和晚期时间点之间几乎没有差异。可以通过重复测量单因素方差分析来跟进问题,发现在性别或早期与晚期之间没有显著差异。
Beta多样性
现在,想使用基于OTU的方法比较各种样本间的成员和结构。开始计算不同样本中成员和结构的相似性。使用dist.shared命令执行此操作,该命令能够将数据稀疏化为相同数量的序列。
mothur>dist.shared(shared=stability.opti_mcc.shared, calc=thetayc-jclass, subsample=t)

这两个距离矩阵(即stability.opti_mcc.jclass.0.03.lt.ave.dist和stability.opti_mcc.thetayc.0.03.lt.ave.dist)可以使用PCoA或NMDS图可视化。主坐标(PCoA)使用基于特征向量的方法以尽可能少的维度表示多维数据。示例数据是高维的(~9维)。
mothur>pcoa(phylip=stability.opti_mcc.thetayc.0.03.lt.ave.dist)

这些命令的输出是以*dist、*pcoa和*pcoa.loadings结尾的三个文件。stability.opti_mcc.thetayc.0.03.lt.ave.pcoa.loadings文件能看到每个轴代表数据总方差的哪一部分。在本次分析中,第一轴和第二轴分别代表总距离变化的45%和14%(59%)。分析输出表明,原始距离矩阵和2D PCoA空间中点之间的距离的R平方为0.89,如果添加第三维,则R平方将增加至0.98。总而言之,还不错。
或者,非度量多维缩放(NMDS)尝试使用用户定义的维数来保持样本之间的距离。可以使用以下命令通过二维NMDS运行数据:
mothur>nmds(phylip=stability.opti_mcc.thetayc.0.03.lt.ave.dist)

打开stability.opti_mcc.thetayc.0.03.lt.ave.nmds.stress文件,可以检查应力和R平方值,这些值描述了排序的质量。该文件中的每一行代表一个不同的迭代,并且在该迭代中获得的具有最低应力的配置记录在stable.opti_mcc.thetayc.0.03.lt.ave.nmds.axes文件中。在此文件中,发现最低应力值为0.11,R平方值为0.95;压力水平(Stress level)实际上还不错。可以通过以下方式测试三个维度会发生什么:
mothur>nmds(phylip=stability.opti_mcc.thetayc.0.03.lt.ave.dist, mindim=3, maxdim=3)

应力值下降到0.05,R平方值上升到0.99。理想的应力值当小于0.20,小于0.10更好。因此,可以得出结论,NMDS比PCoA更好。可以通过绘制stability.opti_mcc.subsample.pick.thetayc.0.03.lt.nmds.axes的内容来绘制NMDS数据的三个维度。同样明显的是,早期和晚期样本彼此分开聚集。归根结底,排序是一种数据可视化工具。可能还想知道在NMDS图中早期和晚期图之间看到的空间分隔是否具有统计显著性,有两个统计工具可供使用。第一个分子变异方差分析(amova),测试代表一组的clouds的中心是否比相同处理的样本之间的差异更分离。这是使用之前创建的距离矩阵完成的,实际上并没有使用排序。可以使用amova命令测试以确定排序内的聚类是否具有统计显着性。要运行amova,首先需要创建一个design文件,指示每个样本属于哪种处理。下载的文件夹中有一个名为mouse.time.design的文件,它看起来像这样:

然后,可以使用此文件运行amova,如下所示:
mothur>amova(phylip=stability.opti_mcc.thetayc.0.03.lt.ave.dist,design=mouse.time.design)

从AMOVA中看到,“cloud”的早期和晚期时间点对于这只老鼠有一个明显不同的质心。因此,在早期和晚期样本中观察到的分离具有统计学意义。还可以使用homova命令查看早期样本中的变化是否与晚期样本中的变化显著不同:
mothur>homova(phylip=stability.opti_mcc.thetayc.0.03.lt.ave.dist,design=mouse.time.design)

可以看到,早期样本的变异量(0.061)大于晚期样本(0.007),差异显著。这就是在最初的研究中发现的——早期的样本比晚期的样本更不稳定。
接下来,想知道哪些OTU将导致样本在两轴间的不同分布。可以通过测量每个OTU的相对丰度与NMDS数据集中的两个轴的相关性来确定这一点。使用corr.axes命令执行此操作:
mothur>corr.axes(axes=stability.opti_mcc.thetayc.0.03.lt.ave.pcoa.axes,shared=stability.opti_mcc.0.03.subsample.shared, method=spearman, numaxes=3)

此命令将生成stability.opti_mcc.0.03.subsample.spearman.corr.axes文件。前五个OTU的数据如下所示:

由于这些OTU所起的作用不同,故以上分析有助于说明OTU对phylotypes的影响。这些数据可以在biplot中绘制,其中从原点(轴1=0,轴2=0,轴3=0)到与每个轴的相关值的辐射线都映射在PCoA或NMDS图的顶部。接下来,使用metastats命令,将看到另一种方法来描述哪些种群造成了特定处理之间的差异。构建biplot的另一种方法是提供指示每个样本的元数据的数据。例如,可能知道这些样本中受试者的体重,身高,血压等。出于讨论目的,提供了mouse.dpw.metadata文件,它看起来像这样:

然后,可以使用metadata选项再次运行corr.axes:
mothur>corr.axes(axes=stability.opti_mcc.thetayc.0.03.lt.ave.pcoa.axes,metadata=mouse.dpw.metadata, method=spearman, numaxes=3)

打开文件mouse.dpw.spearman.corr.axes可以看到:

表示随着断乳后时间的延长,群落将沿着轴3向正方向移动。
可以使用的另一个工具是get.communitytype,以查看数据是否可以划分为不同的群落类型。
mothur>get.communitytype(shared=stability.opti_mcc.0.03.subsample.shared)

可以看到最小的Laplace值适用于K值为2(10643.60)。这表明样本属于两种群落类型。打开stability.opti_mcc.0.03.subsample.0.03.dmm.mix.design,可以看到所有后期样本和Day0样本都属于Partition_1,而其他早期样本则属于Partition_2。可以查看stability.opti_mcc.0.03.subsample.0.03.dmm.mix.summary文件,以了解哪些OTUs最能代表群落差异:
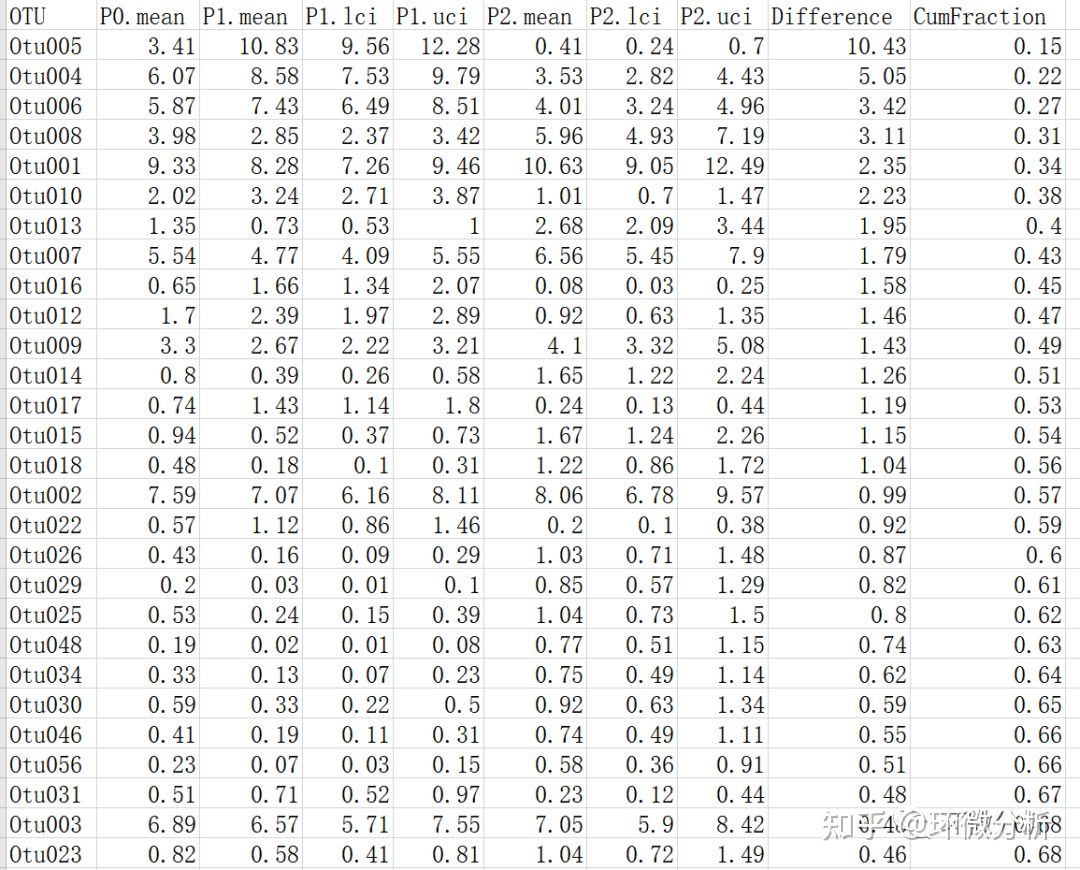
还可以将这些OTU标签与stability.opti_mcc.cons.taxonomy文件中的一致分类进行交叉引用,以获得这些生物的名称。
Population-level analysis
除了使用corr.axes和get.communitytype,还有几种工具可以区分不同的样本组。演示的第一个参数是metastats,它是一个非参数T检验,用于确定本研究中雌雄样本之间是否存在差异表示的任何OTU。在Mothur中运行以下命令:
mothur>metastats(shared=stability.opti_mcc.0.03.subsample.shared,design=mouse.time.design)

从stability.opti_mcc.0.03.subsample.0.03.Late-Early.metastats文件中查看前五个OTU,我们看到以下内容...

这些数据说明,早期样本和晚期样本之间的OTU4、5和6显著不同。
可以用来替代metastats的另一个非参数工具是lefse:
mothur>lefse(shared=stability.opti_mcc.0.03.subsample.shared, design=mouse.time.design)

Number of significantly discriminative features:90(94)before internal wilcoxon.
Number of discriminative features with abs LDA score>2:90.
在lefse_summary文件的顶部,可以看到:

两组之间的OTU4、5和6显著不同,并且在后期样本中显著升高。
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!














 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









