






目的:受体和配体柔性均考虑的分子对接
所需功能模块:Discovery Studio Visualizer client,DS LibDock,DS Catalyst Conformation,DS CHARMm及DS Protein Refine
所需数据文件:1s1x_prot.dsv和1fk9_lig.sd
所需时间:25min
介绍
柔性分子对接是指在对接过程中,蛋白的侧链和配体分子构象都可以自由变化,多用于精确考查分子间的结合模式,但由于计算量和计算时间的缘故,在实际应用中,一般只将活性位点处残基侧链定义为柔性,可以考虑其构象的改变。在Flexible Docking中,配体将按如下步骤对接到考虑活性部位氨基酸残基侧链柔性的受体中:
- 选择受体柔性氨基酸残基,使用ChiFlex(CHARMm)通过改变侧链构象计算出一组蛋白构象
- 使用LibDock将柔性配体对接到每一个受体构象的活性部位
- 使用ChiRotor(CHARMm)在刚性配体存在的情况下优化选定的蛋白侧链
- 使用CDOCKER优化最后的配体对接构象
在本教程中研究的两个蛋白分子为同一个HIV-RT受体,但由于所含的配体不同,两蛋白PDB编号不同,分别为1s1x和1fk9。配体结构的差别会引起蛋白构象的改变,这也是柔性对接的理论基础。本教程将1fk9中结合的配体对接到1s1x中。
具体步骤包括:
- 准备对接的分子体系
- 执行分子对接计算
- 分析对接结果
- 计算对接结果的RMSD值
准备对接的分子体系
1.蛋白和配体文件的准备
在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions| 1s1x_prot.dsv文件。
该蛋白将在一个新的三维窗口中打开。(图1)
此蛋白文件已经过处理:已赋予CHARMm力场,所以在对接之前不需再进行力场的赋予;此外,结合位点也已识别,窗口中只显示该结合位点周围的8个残基及其相应的残基名,在对接过程中将会考虑这8个残基的侧链柔性,上述8个残基已被定义为一组并命名为1s1x_res8,系统视图中已显示。
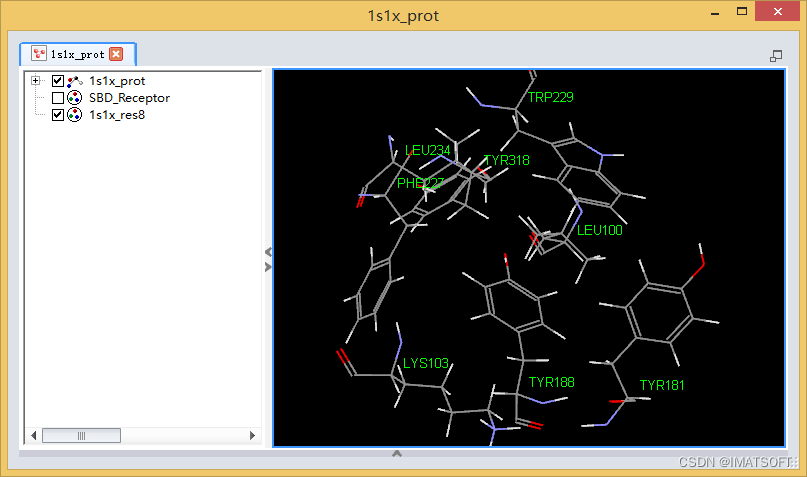
图1 打开蛋白文件
在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions|1fk9_lig.sd文件。
在一个新的窗口中打开配体分子,默认以表格视图形式显示。
按Ctrl+G,展开分子视图窗口,显示配体分子结构(图2)。
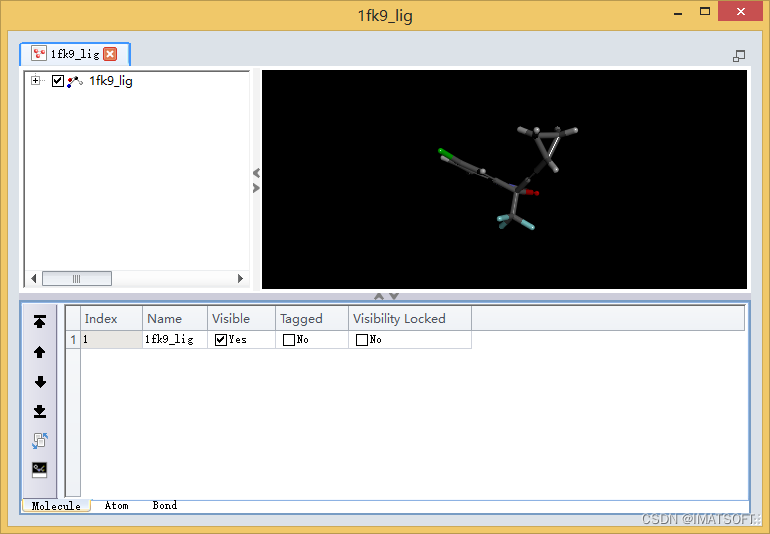
图2 打开配体分子
执行分子对接计算
在流程浏览器(Protocols Explorer)中,展开Receptor-Ligand Interactions | Docking ,点击打开Flexible Docking柔性对接流程。
流程的对应参数项在参数浏览器中出现。
在参数浏览器中,点击Input Typed Protein Molecule参数,从下拉列表中选择1s1x_prot:1s1x_prot设置受体蛋白。
点击Input Ligands参数,从下拉列表中选择1fk9_lig:All,设置配体分子。
点击Input Site Sphere参数项,从下拉列表中选择binding_Sphere坐标,指定活性位点。
点击Selected Residues参数项,从下拉列表中选择1s1x_res8,指定考虑侧链柔性的一组氨基酸残基 。
该操作选择了蛋白活性位点处残基。这些残基事先已被选中并被定义为1s1x_res8组。
点击Generate Protein Conformations参数项,选择False。
展开Generate Protein Conformations参数组,点击Input File参数项,载入之前已经产生好的蛋白多构象。在文件浏览器(Files Explorer)中,找到Samples| Tutorials| Receptor-Ligand Interactions| 1s1x_prot-conformations.mol2文件,载入即可。
本教程为了缩短运行时间,蛋白多构象已事先产生好。
展开Generate Ligand Conformations参数组,点击Conformation Method参数,从下拉列表中选择BEST方法。
注:虽然BEST方法不是构象生成方法参数缺省设置,本例中使用该方法是因为对于含复杂环的配体它能更好的覆盖构象空间。
展开Docking参数组,点击参数Max Hits to Save设置最多保存结果为3。
如果电脑配置有多个核,则可以设置并行设置来缩短运行时间。
点击Parallel Processing参数,设置为True。(图3)
点击流程工具栏(Protocols toolbar)中的Run![]() 按钮运行作业,等待作业完成。
按钮运行作业,等待作业完成。
当作业完成时会出现作业完成对话框,关闭作业完成对话框。
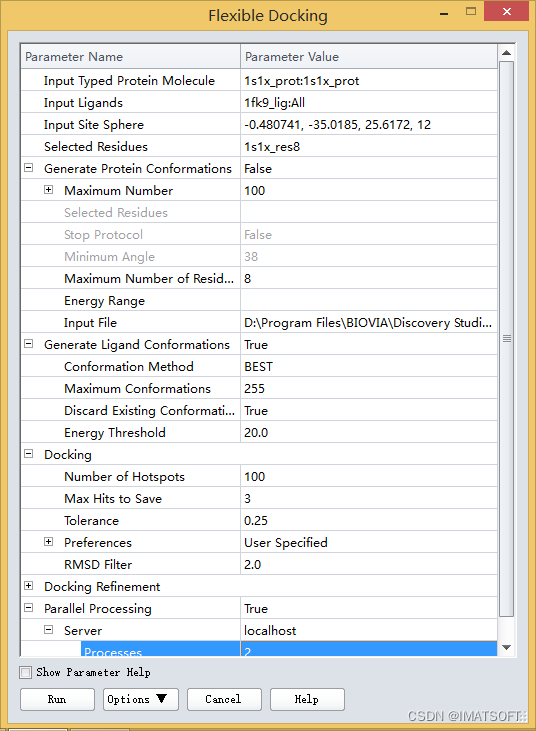
图3 “Flexible Docking”参数设置
分析分子对接结果
在开始结果浏览之前,为了清晰起见,在菜单栏中选择Window | Close All关闭所有窗口。
在作业浏览器(Jobs Explorer)中,双击刚完成的作业。
在Html窗口中打开Report.htm文件。
在Html窗口中,点击Results部分的View Results。
打开一个新的分子窗口,显示1s1x_prot.dsv受体分子和1fk9_lig.sd文件(配体的对接构象)。
在表格视图中,将第一个1fk9_lig对接构象的visible勾选上,让其显示在分子视图窗口中。(图4)
然后点击表格中的![]() 键和
键和![]() 键浏览对接得到的各配体构象。
键浏览对接得到的各配体构象。
观察1s1x_res8组定义的氨基酸残基侧链构象将随着配体构象的变化而变化。
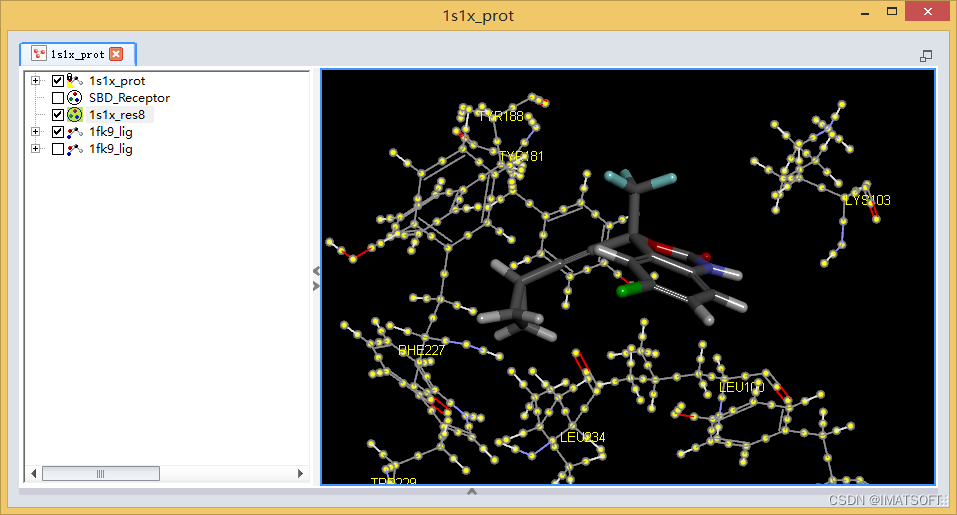
图4 对接结果示意图
从表格中还可以查看每个对接构象的对接打分,包括CDOCKER的能量打分,也包括LibDockScore得分。
从菜单栏中选择Window | Close All,关闭所有窗口。
计算对接构象的RMSD值
下面我们计算配体对接构象同晶体构象的RMSD值并分析其CDOCKER能量。
在任务浏览器(Jobs Explorer)中,单击该任务条下(点开前面的+号)Flexible Docking链接。点击Docked Ligands.

对接配体将在一个新的分子窗口中打开。
按住CTRL+G,显示分子图形窗口。
展开菜单栏File | Insert From,点击File...,选择Samples | Tutorials | Receptor-Ligand Interactions | 1fk9_lig.sd。
在同一窗口中插入1fk9_lig的天然晶体结构。
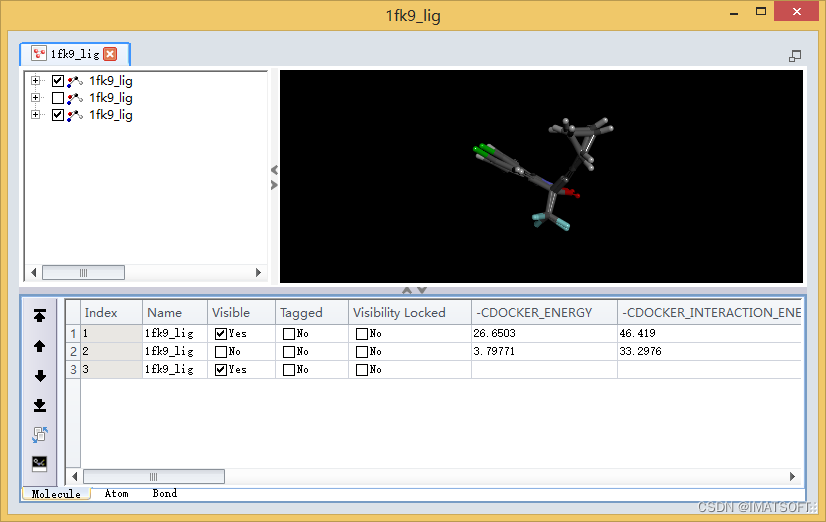
图5 配体晶体结构与对接结果叠合
在表格视图中,选择最后一行的1fk9_lig分子,展开菜单栏Structure|RMSD,点击Set Reference。
在分子窗口中点击鼠标右键,选择Show All,显示所有小分子结构。
展开菜单栏Structure|RMSD,点击Heavy Atoms。
打开一个新的窗口,显示所有2个对接构象同晶体构象之间的RMSD偏差。

图6















 4976
4976











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









