






1、PDB结构下载:在PDB官网下载靶点的蛋白结构,筛选条件:人源,X-ray,分辨率在3.0以下
2、筛选蛋白结构:没有复合物的不要,只保留一个小分子和一条链,其他操作:去水或者其他杂原子选做
3、做一个分子重合,quick align,看看蛋白质结构重合度
4、蛋白质准备:找到对接的最好构象并保存成maestro格式(.mae)(可以使用ome step protein preparation批量处理蛋白)
5、导出小分子,然后做配体准备,导出文件格式为SDF(.sdf)
6、开始对接,用cross docking 模块对接,受体选择复合物(complex)将保存好的复合物文件上传,将配体文件也上传,(可以更改一些设置,例如用几个核运行和分成几个任务),点击运行
注:这个这是一般流程·,具体操作要看实际情况。
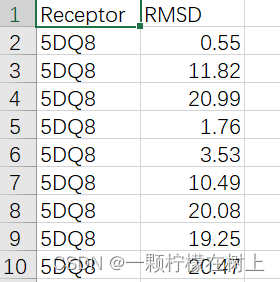
后续处理:因为交叉对接是选择最好的PDB蛋白质构象,后面交叉对接后需要处理数据,在薛定谔的log输出里面找到Receptor 和RMSD值,把数据放入csv文件中,然后用一个python代码进行数据处理。
import pandas as pd
import math
data= pd.read_csv('D:/desk/rmsd.csv')
value_mean = data.groupby(['Receptor'],as_index = False,group_keys=False)['RMSD'].mean()
value_std = data.groupby(['Receptor'],as_index = False,group_keys=False)['RMSD'].std()
value_lower_than_2 = data.loc[data['RMSD']<=2]
rmsd_lower_than_2_count = value_lower_than_2.groupby(['Receptor'],as_index = False,group_keys=False)['RMSD'].count()
result = pd.concat([value_mean, value_std,rmsd_lower_than_2_count], axis=1)
print(result)
result.to_csv('result1.csv',index=False)RMSD<=2的蛋白质结构比较适合

















 8587
8587

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









