







本篇文章初稿由组内pf同学倾情整理,感谢大神奉献!@BlastOrange
25.03.19回顾时发现漏洞和需要补充的地方。二次修正
呃呃呃呃修订了好多,结果CSDN卡住退出了……痛
需要大家自行准备多个蛋白质(PDB格式)和多个小分子配体(最好是mol2格式)。
注意,准备的蛋白质最好有共晶配体,没有的话没关系,见4.缺少配体/配体识别错误的蛋白处理。
在开始批量对接前,确保你
- 设置好了薛定谔的工作目录。
- 在工作目录中新建了两个文件夹,分别是
prep_protein来存放处理好的蛋白,prep_lig来存放处理好的配体。
1.处理蛋白质
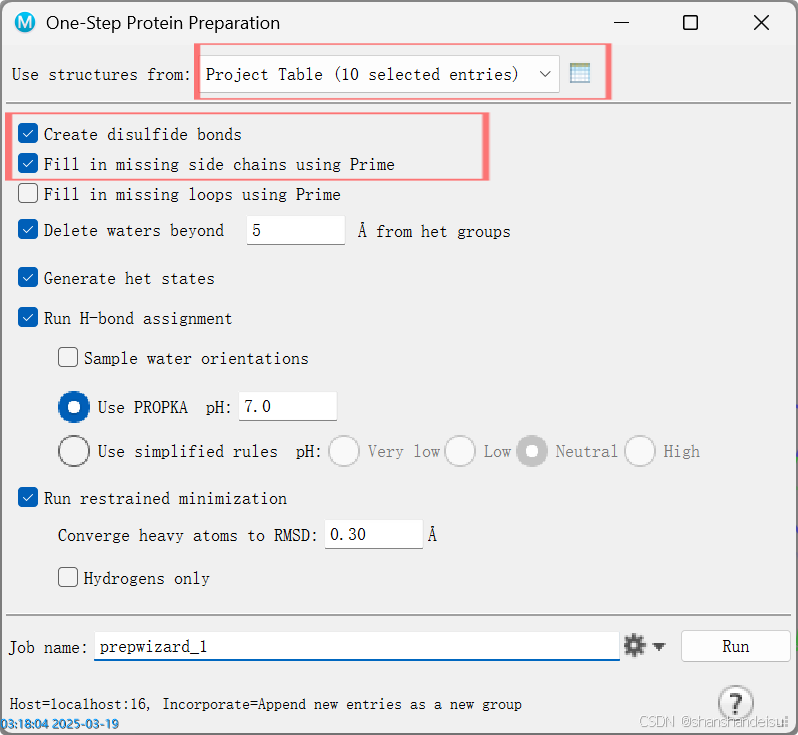
导入蛋白质分子到薛定谔中,在工作区中选中这些蛋白,打开one step Protein Preperations模块。勾选:
Create disulfide bonds(如有二硫键,补全二硫键)Fill in missing side chains using Prime(补全残基)
job settings设置好后点击RUN运行即可,其余参数在本人其他博客均有提及。
也可以不设置
job settings,但可能会拖慢速度,设置时任务顶多跑几分钟左右。不设置可能会十几分钟。
任务完成后,选中所有处理好的蛋白,右键点击export_Structures输出结构到prep_protein文件夹中。
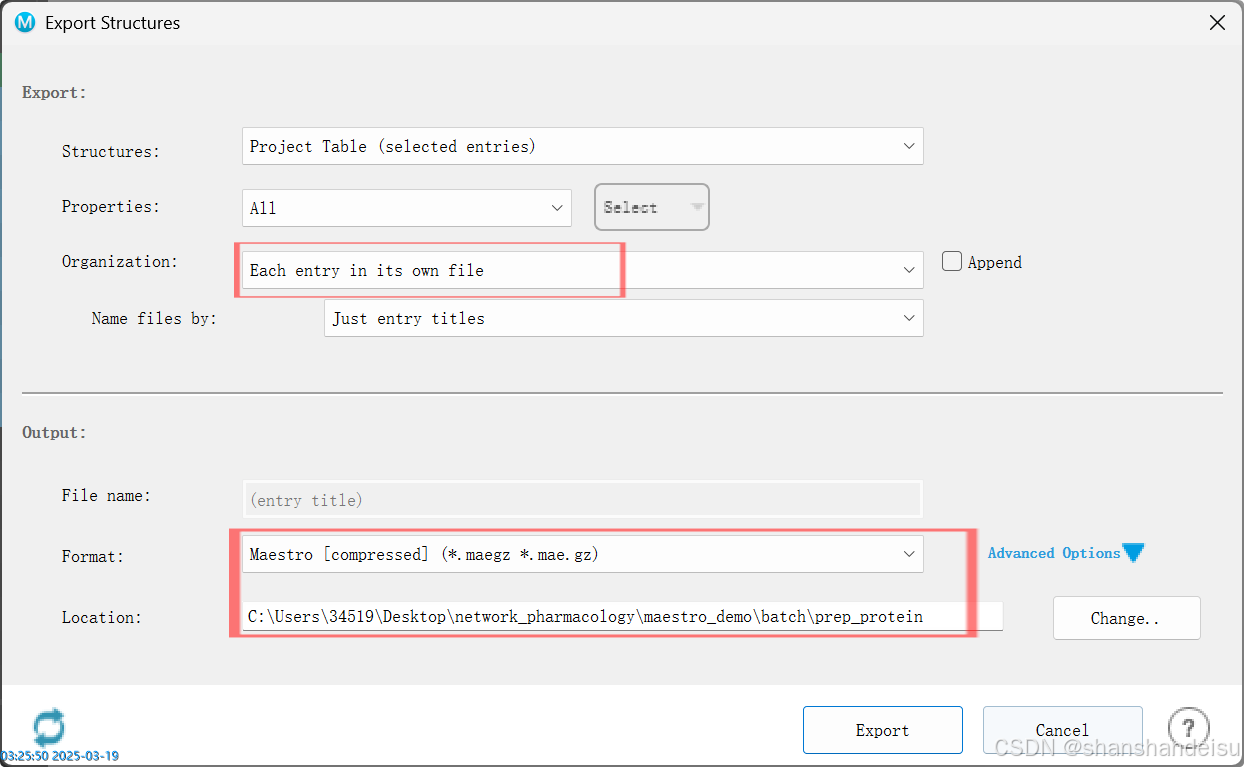
.mae和.maegz没什么区别,只是后者采用算法压缩,占用空间更少,批量操作时更推荐.maegz格式。
2.处理配体
接着导入小分子配体到薛定谔中,在工作区选中所有小分子,找到LigPrep模块。
注意勾选 Determin chiralities from 3D structure(如果你的配体是mol2格式的话,不是的话见本人单个对接的博客)。
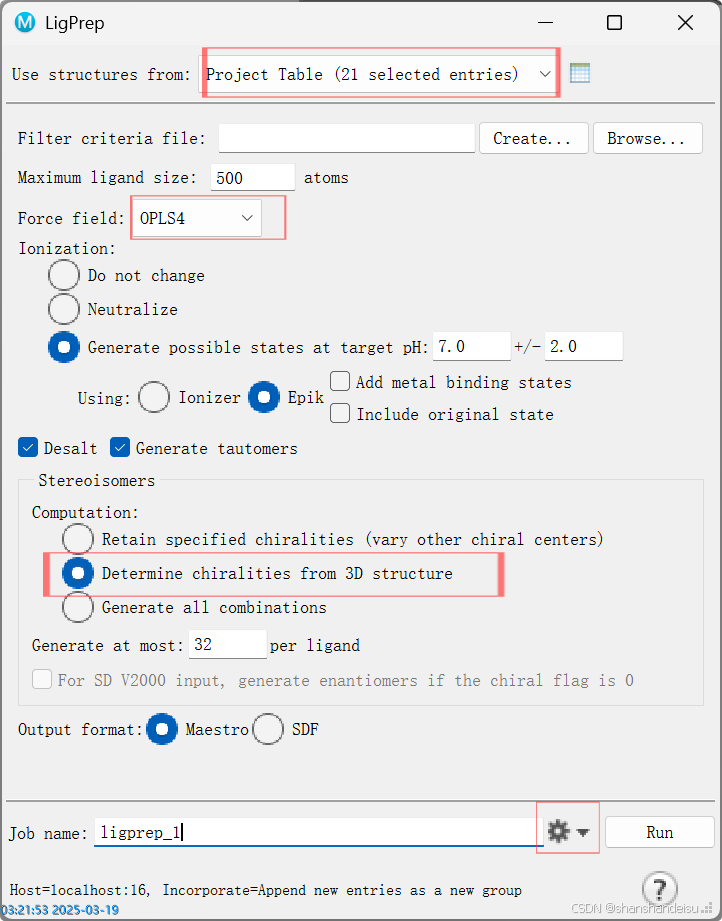
不建议使用OPSL2005,理由如下:
| 特性 | OPLS4 | OPLS_2005 |
|---|---|---|
| 发布时间 | 2018年(更新) | 2005年(较早版本) |
| 参数覆盖范围 | 更广,覆盖更多官能团和药物分子 | 基础覆盖,适合常见有机分子 |
| 精度验证 | 基于量子力学和大规模实验数据优化 | 基于经典力场参数化 |
| 分子对接表现 | 更准确的结合模式预测和打分 | 适用于一般性对接场景 |
| 计算效率 | 稍高(因参数更复杂) | 略快(参数简化) |
| 特殊结构支持 | 支持金属配合物、大环化合物等复杂结构 | 对非标准结构处理有限 |
任务完成后,选中所有处理好的配体,右键点击export_Structures输出结构。
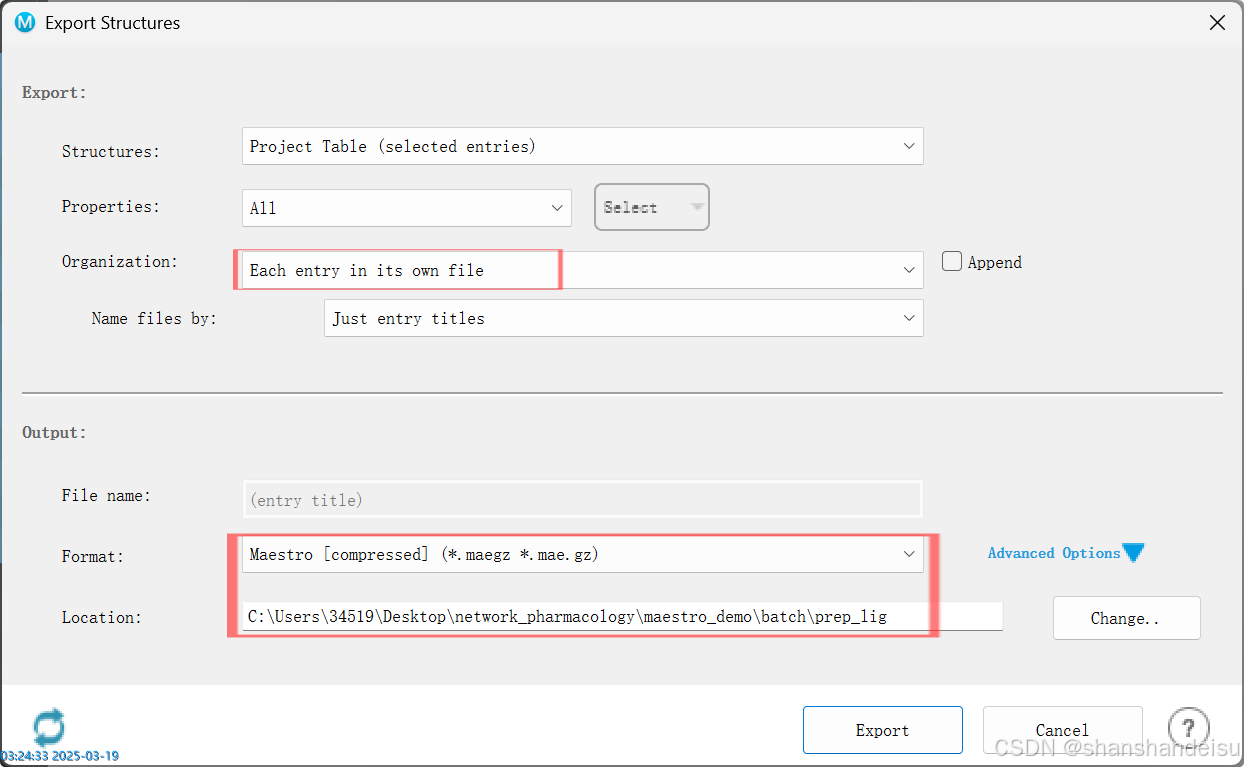
此时工作区可以删除配体条目,避免工作区太多条目。
3.批量对接
3.1.输入蛋白
找到Crossdocking模块(原名xGlide)。
点击Add Complex…选择prep_protein的蛋白质文件。
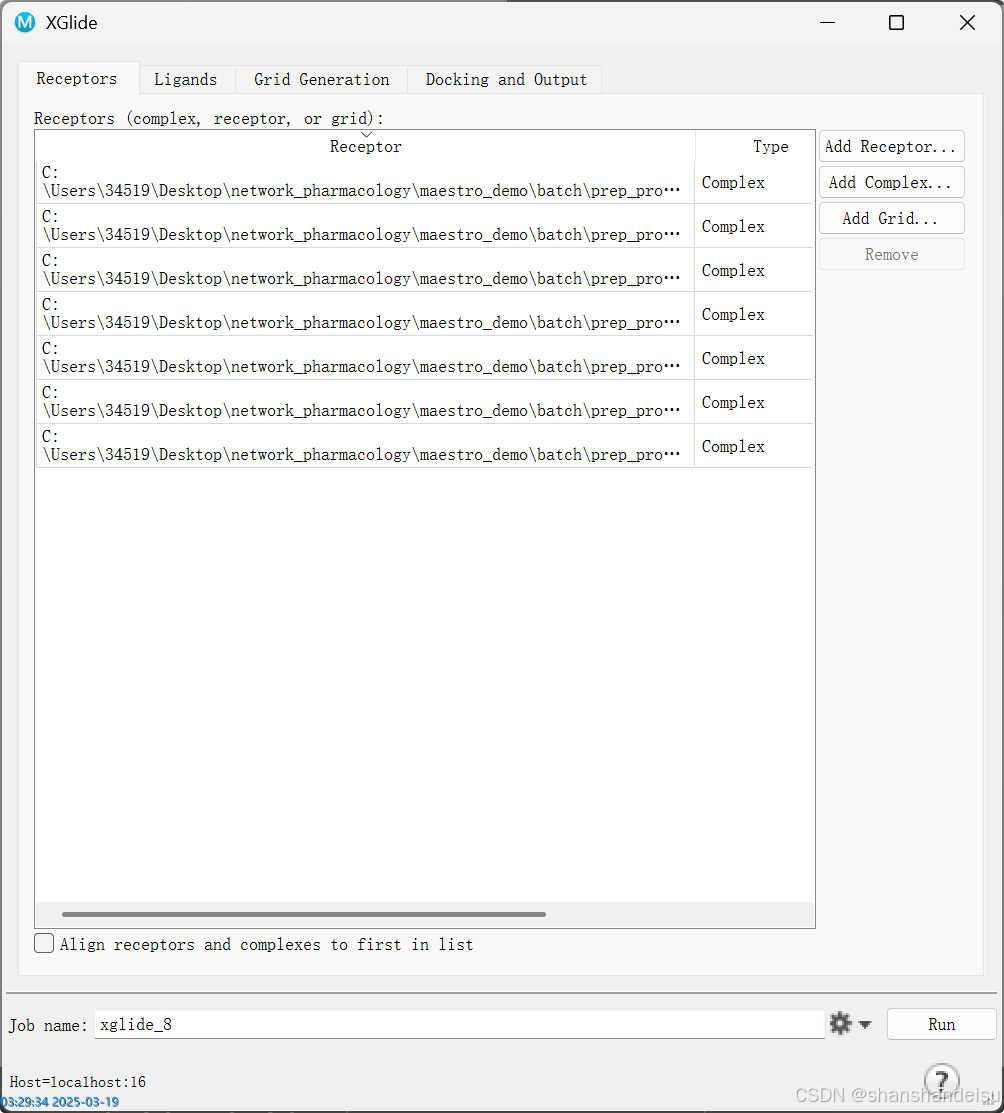
可以看到,在这我们输入了7个蛋白。
3.1.1.弹出窗口:Choose Ligand
如果弹出窗口如下,说明你的蛋白中含有配体2个或2个以上。
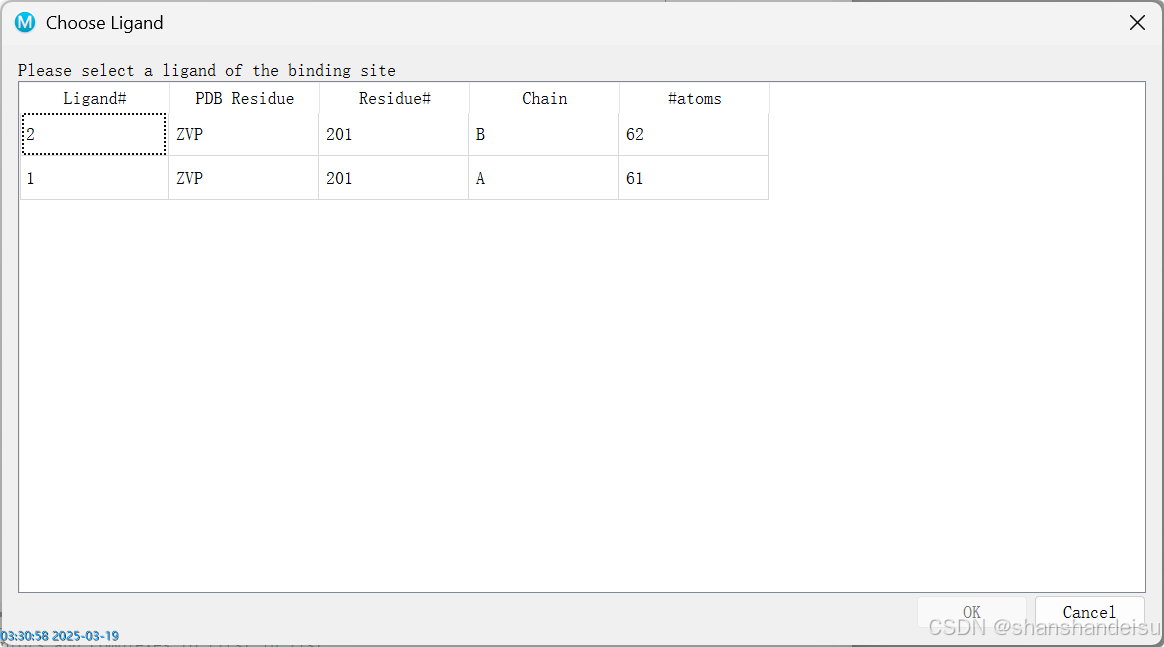
本人是建议随便选一个即可。因为这个配体是蛋白原本含有的配体,作用只是在最后对接结果中会用原来的配体也对接一次,作为阳性对照,输出在第一行。
3.1.2.报错:Speicified structure did not have any ligands
字面意思,你的蛋白中不含有配体。 解决方案见4.缺少配体/配体识别错误的蛋白处理
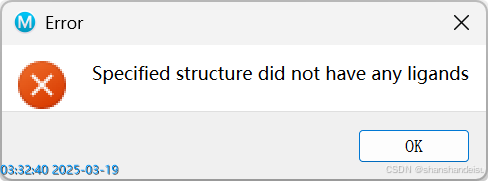
其实我觉得这是薛定谔软件做的不到位的地方,按理来说我是批量输入蛋白的,其中一般只会有几个是不含有配体的蛋白,它应该至少报错告诉我是哪几个蛋白不含有配体,我好方便额外处理。并且把含有配体的蛋白正常输入。
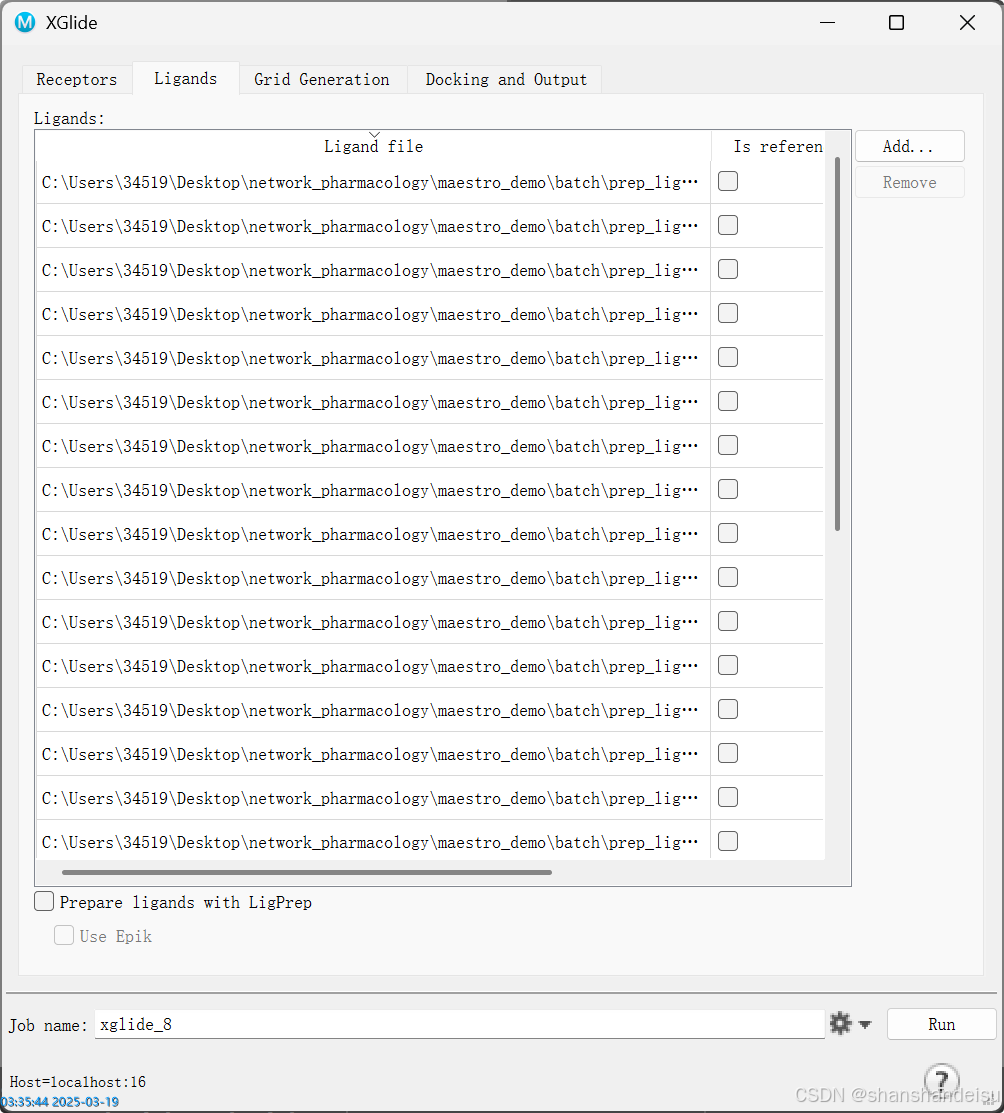
3.2.输入配体
切换到ligand标签页,同样全选prep_lig中的所有配体。
可以看到这里我们输入了21个配体进行对接。
3.3.对接设置
点击Gird Generation,选择第二项,job settings设置好后点击RUN即可。
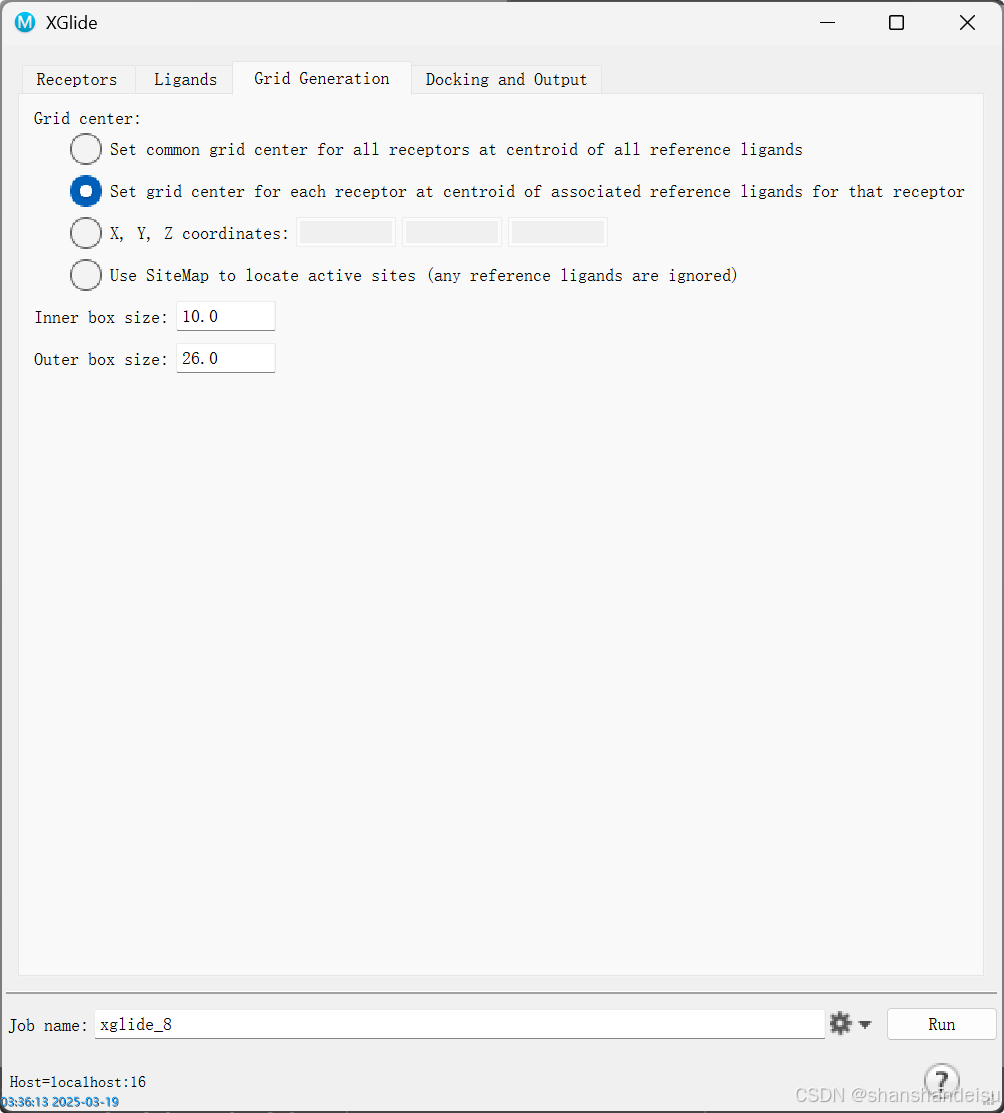
选项第一个:生成一个口袋,所有配体共用一个口袋(即一对多对接)
选项第二个:为每个配体生成一个单独的口袋用来对接(即交叉对接)
如果你的配体较大,可以调大outer boxsize,不过一般默认即可。
3.4.查看结果
对接完成后打开xglide_1文件夹(我这因为对接了很多次,所以打开了xglide_7),显示如下:
可以直接将里面所有的.maegz文件拖入软件查看结果。
或者file/import structure打开xglide_1文件夹,选择所有.maegz文件。
总之,工作区出现如下7个条目,分别对应7个蛋白:
点击右上角table即可查看对接分数。
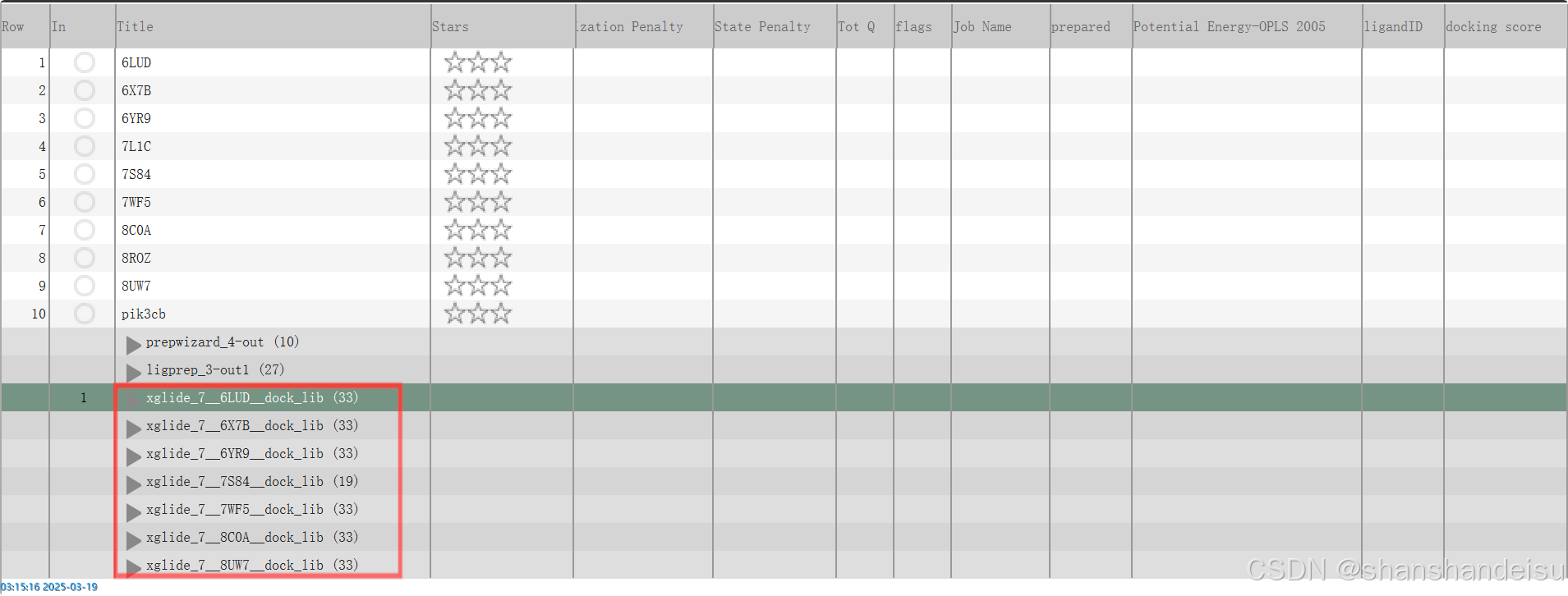
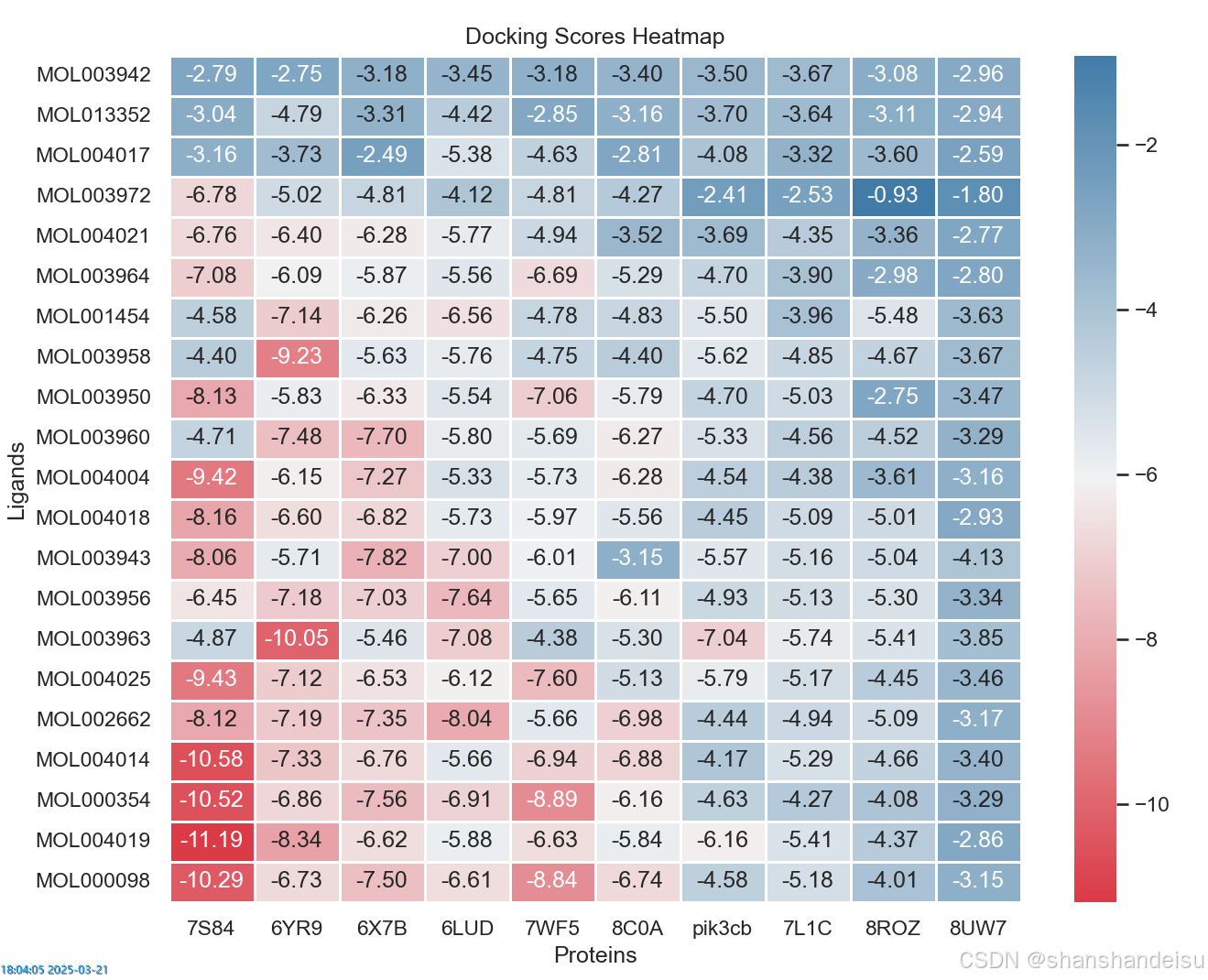
其实可以看到,效果还是很好的,基本就差阳性对照零点几分(骄傲(╹ڡ╹ ),普遍都是-7左右。
3.4.1.很好用的小技巧1:标注结果
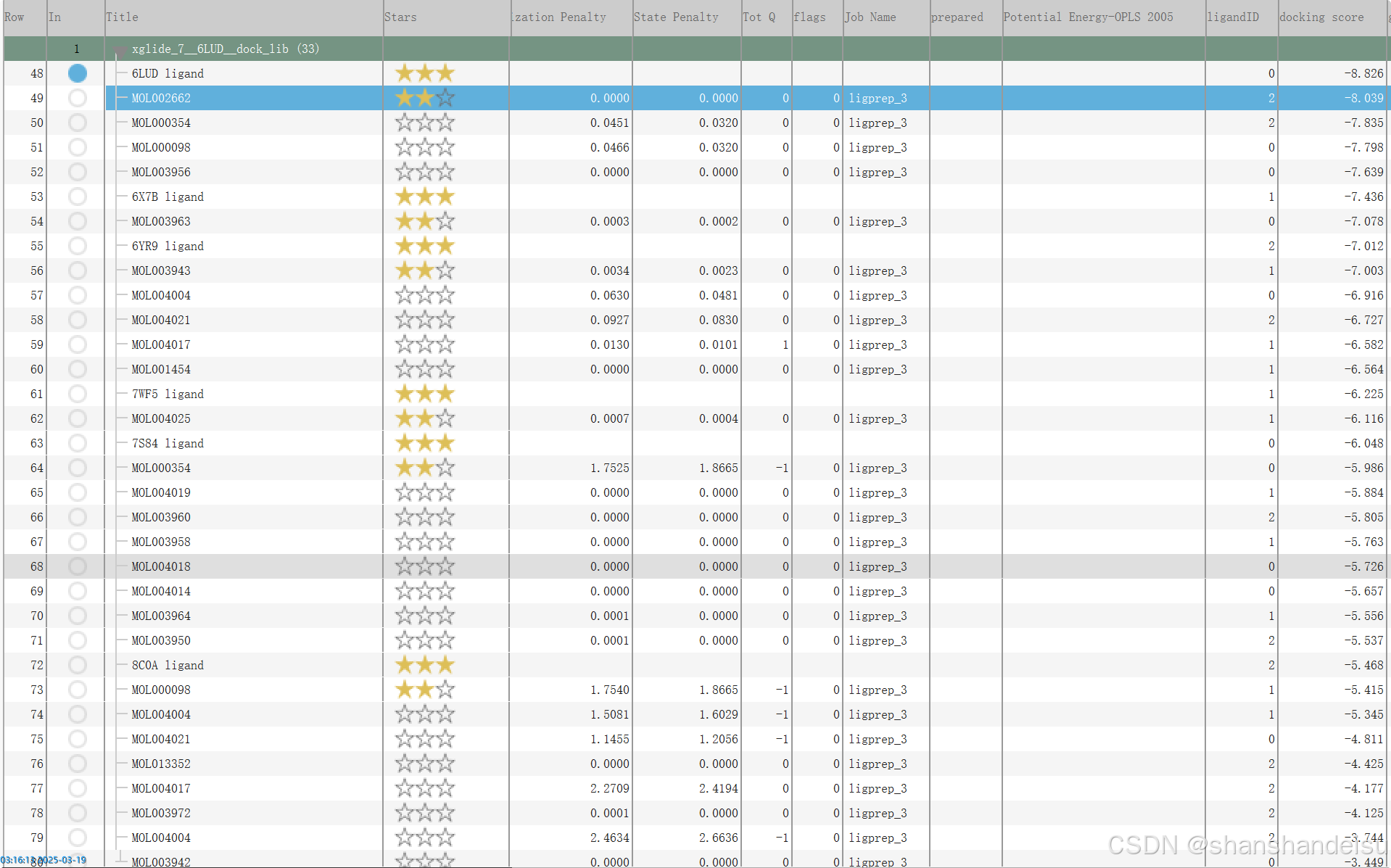
在这我们以6LUD蛋白为例,我们点击星星可以手动标注自己满意的结果。
以PDB_ID Ligand为名的,就是蛋白本身含有的配体对接出来的结果,作为阳性对照。所以我个人会习惯给他们打上三颗星标注清楚。
因为条目是按照对接分数排名的,所以在这些阳性对照下面一般就是该蛋白对接分数最高的配体条目。我会标注上两颗星。
3.4.2.很好用的小技巧2:只展示对接分数
可以看到,这个表格展示的属性非常多,而且还有其他条目,但是我们只想看对接结果的对接分数怎么办?
实际上在表格的最右边存在一个属性树,我们可以只勾选Glide/Primary/docking score和Maestro/Source File
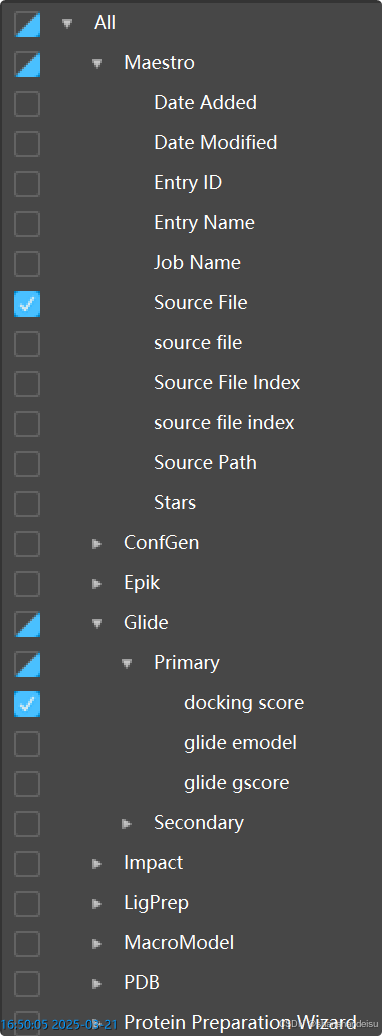
现在的表格界面会变得干净很多,只剩下对接分数和源文件。
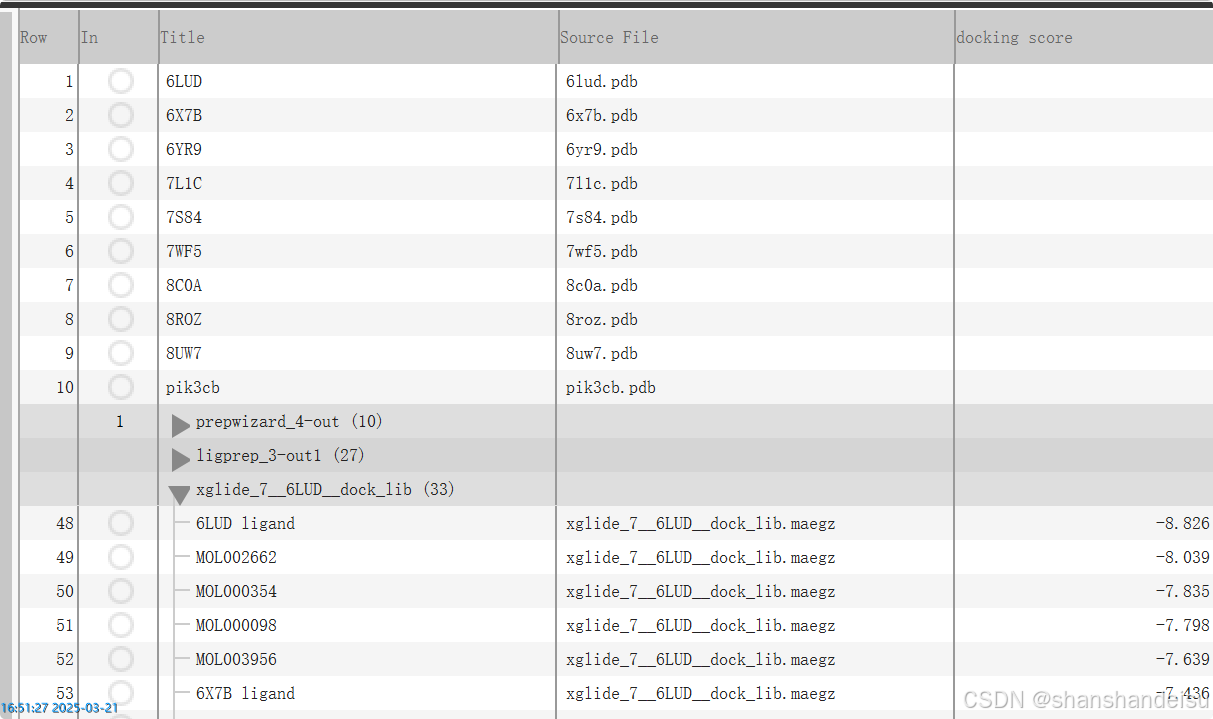
如果你在这个页面最右边没有找到属性树,说明导航栏忘记打开它了。
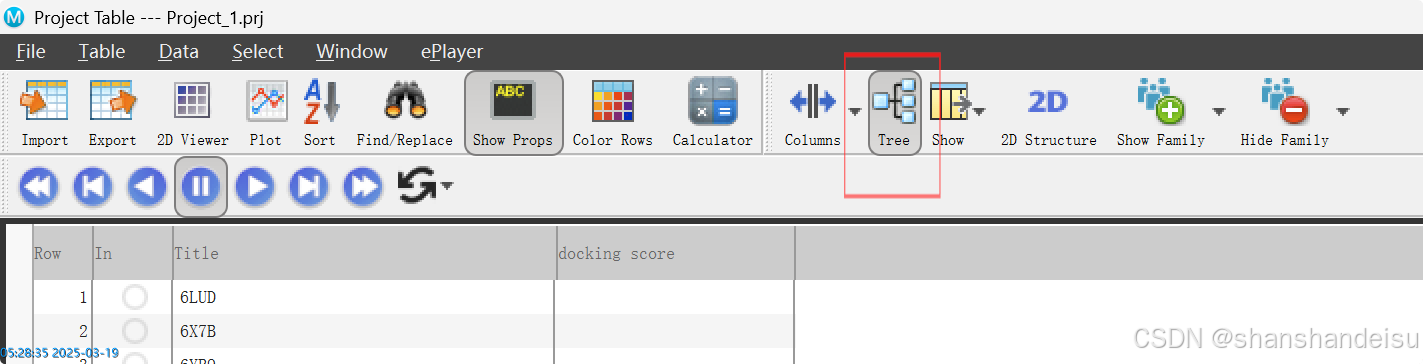
3.4.3.处理表格
最后在顶端导航栏导出表格Data/Export/Spreedsheet,注意勾选selected,保留3.4.3.我们提到的信息,并且以制表符分隔。
最后表格如下:
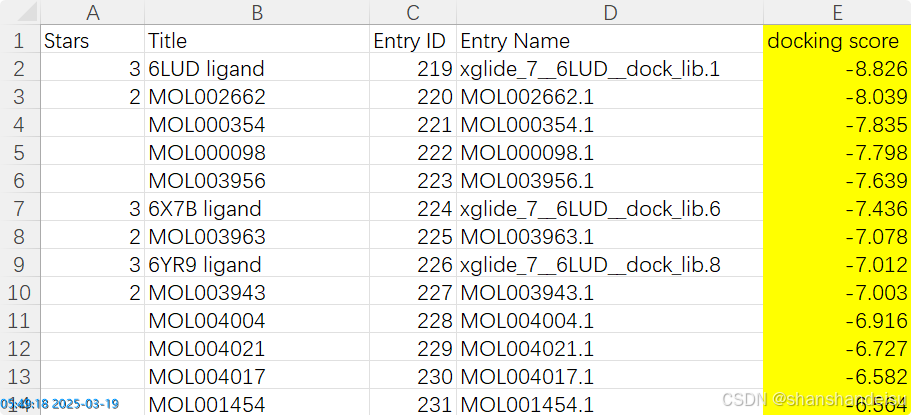
如果有更好的导出表格的方式欢迎评论区补充。
然后我们需要进行表格处理。
首先是生成一份含有共晶配体对比的数据表格,自己方便比较数据。
然后是生成一份不含共晶配体对比的数据表格,主要用来生成python热图。
dddd代码太长了,此处省略
总之,我们得到了这两个表格,打开含有共晶配体的表格来看下:
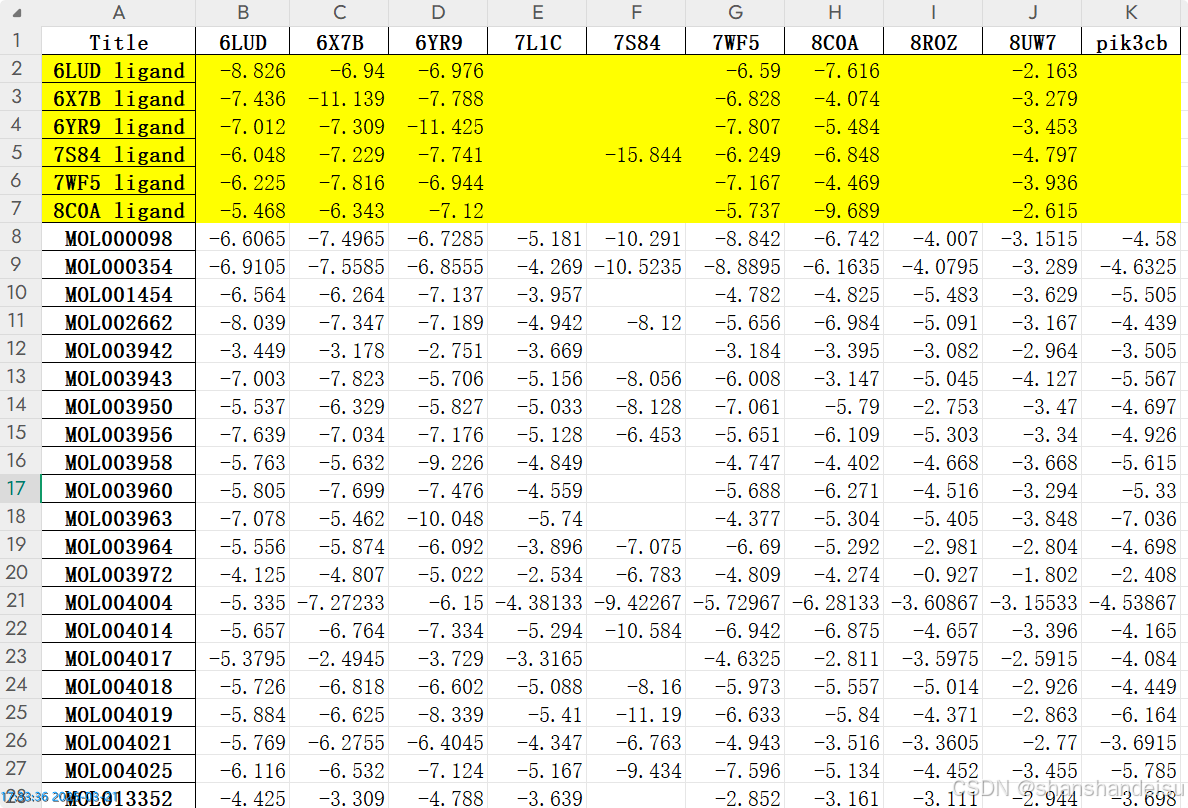
可以看到,标黄的就是共晶配体的对接分数。
为什么不用这一份生成热图,其实也是可以的,只不过我还处理了三个不含配体的蛋白,所以会有很多地方数据是缺的。
如果你数据不缺的话,可以直接用这一个表格跑热图。
除此之外,还有一些空缺的数据,这里主要是7S84这个蛋白,可能是对接的结果不是很好,总之需要单独再对他跑一次(推荐这次用SiteMap来做,具体见4.缺少配体/配体识别的蛋白处理,或者你可以换成XP来做)。
我这里单独跑完看了下数据:
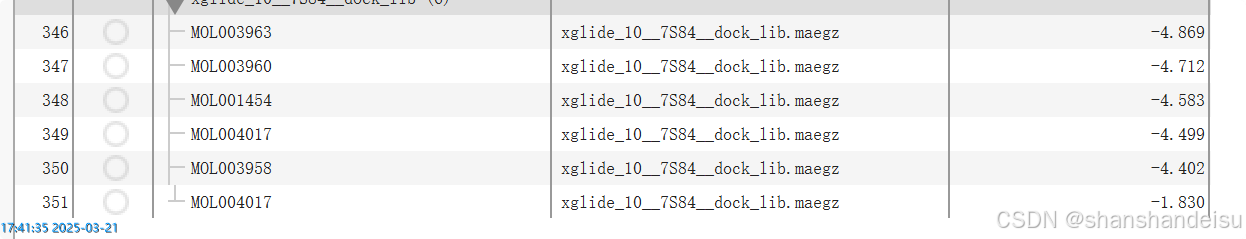
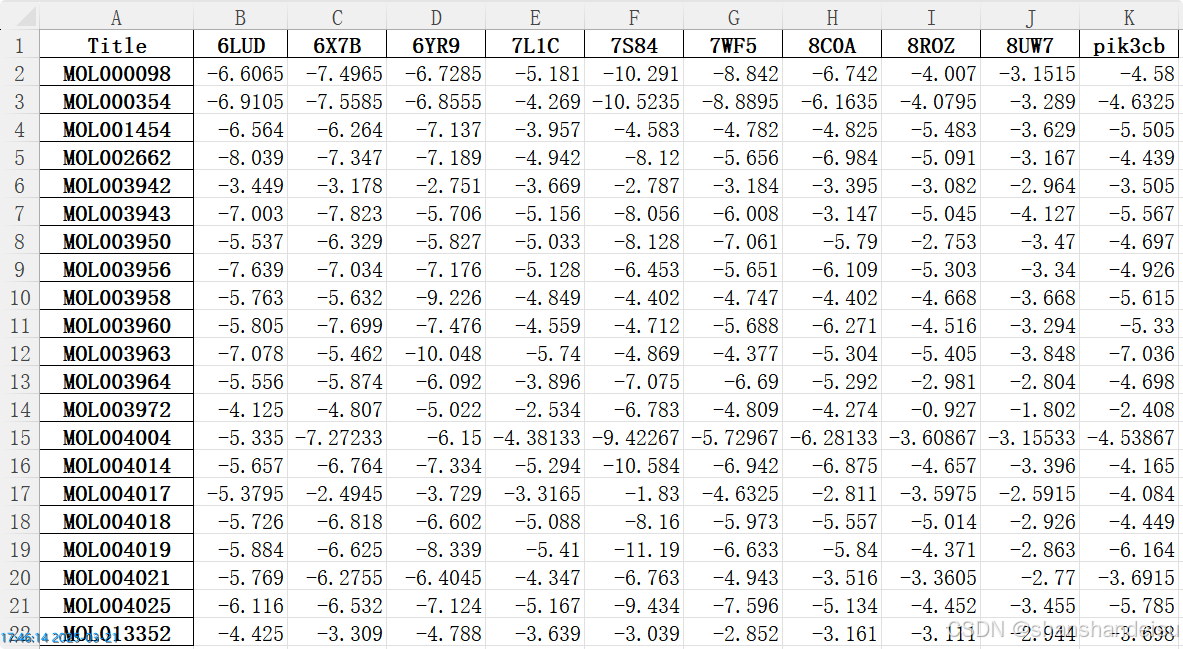
确实都是-4点多,数据都不太好,总之最后我们补全了数据,拿到的不含共晶配体的表格如下:
3.4.4.python生成热图
dddd代码太长了,省略
结果如图:
4.缺少配体/配体识别错误的蛋白处理(SiteMap口袋识别)
除了以上正常的蛋白中含有共晶配体的情况,你可能还存在以下情况:
- 蛋白确实没有共晶配体
- 蛋白有共晶配体,但薛定谔识别错误。
在这我分别举例两个蛋白。首先是PIK3CB,这个蛋白在PDB数据库找不到,所以我是用alphafold3预测了结构,所以其没有共晶配体。
其次就是PDB ID为7L1C的蛋白,它在PDB数据库如下:
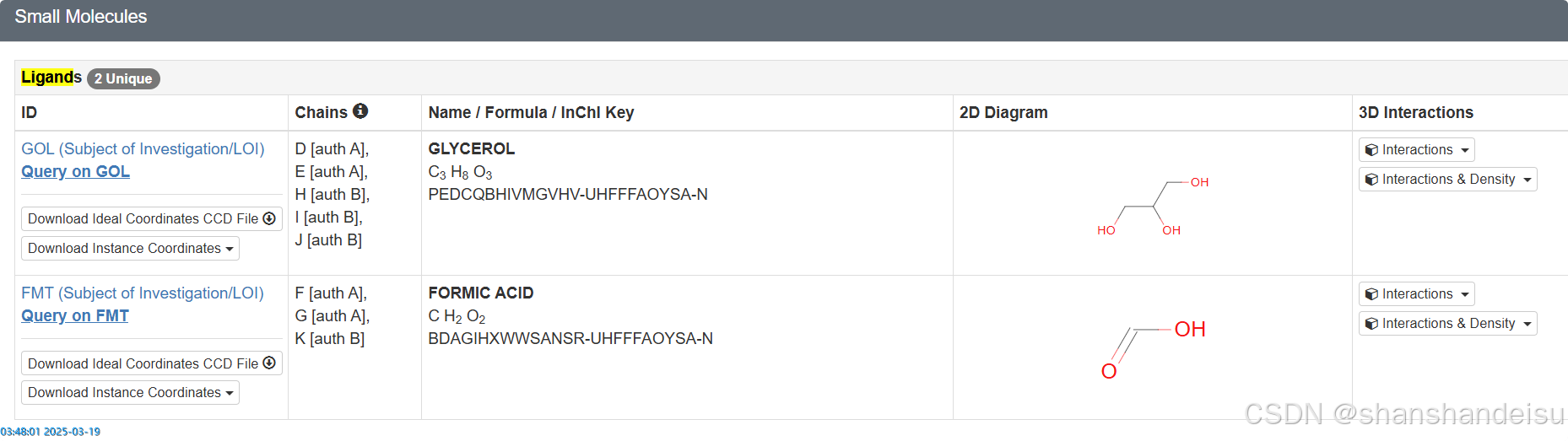
可以看到确实存在两个共晶配体的。然而其放入薛定谔后,识别的层级目录如下:
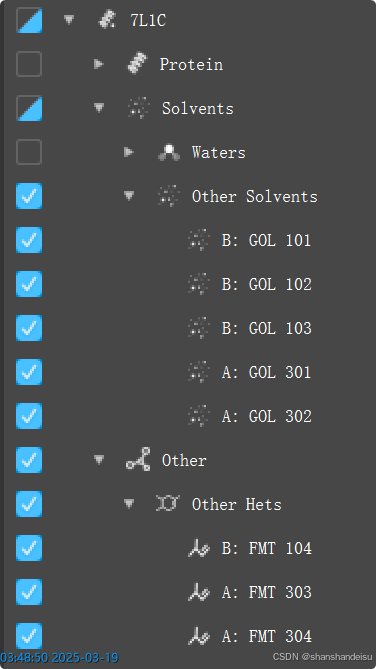
可以看到没有被识别成Ligand层。
对以上情况蛋白的解决方案,可以很简单。我这边有三四个方案hhh,在下文我说我最推荐的一种。
其实为什么要在第一个标签页就区分add complex还是add receptor,就是因为薛定谔想在这一步就根据你的输入来提前判断,如果你选择的是add complex,那么它就默认你一定会用上蛋白中自带的配体,所以才会报错。
所以我们在3.1.输入蛋白时,选择Add Receptor,不再选择Add Complex即可。
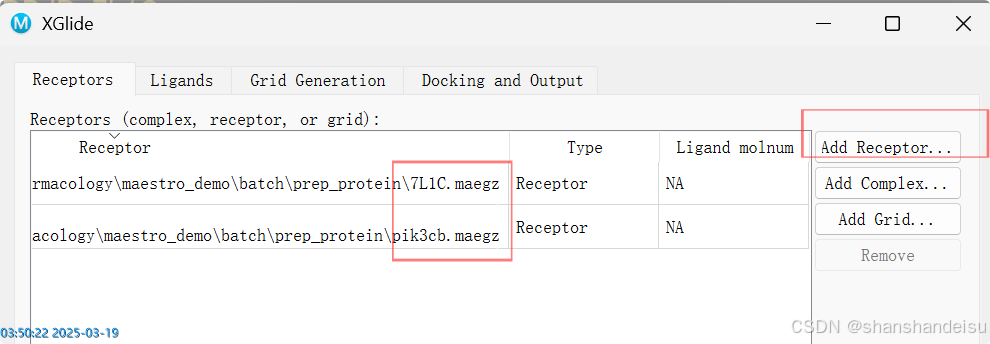
然后我们可以在3.3.对接设置时,选择第四个选项Use SiteMap,使用薛定谔自己的口袋识别算法,自动识别蛋白中的口袋(这个模块我单个对接的博客也有讲到。
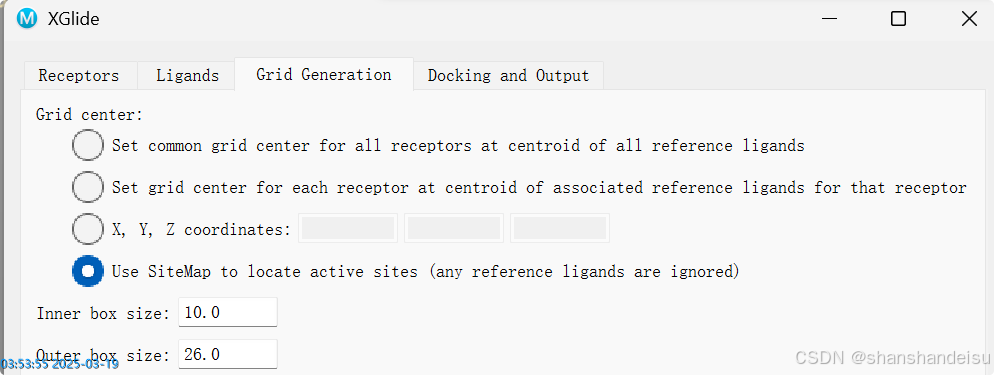
其他步骤和前面一样。不过这个方案用时会比第二个选项要慢,毕竟要多一步预测嘛。
除此之外,你也可以使用选项第三个进行盲对接。
最后的最后,对于含有配体的蛋白,实际上你也可以在第1步处理蛋白的时候就把它们的共晶配体全部去掉,这样就可以和缺少配体的蛋白同批次用sitemap处理啦。
好处就是全部合并为一个批次。
坏处就是要手动去除下配体(直接层级结构去除都行),然后对接时间会长一点。
准确度目测是不会差很多的。



















 8573
8573

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











