







 超级会员免费看
超级会员免费看
使用bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP变异位点数量统计
软件安装
1. conda 安装
conda install bedtools -y
2. 源码安装
wget https://github.com/arq5x/bedtools2/releases/download/v2.28.0/bedtools-2.28.0.tar.gz
tar -zxvf bedtools-2.28.0.tar.gz
cd bedtools2
make
1. 准备染色体大小文件
# 安装python库
pip install pyfaidx
# 使用faidx获取hg19参考基因组序列每条染色体的大小
faidx hg19.fa -i chromsizes > size.genome
# 获取不包含线粒体chrM的大小文件
faidx hg19.fa -i chromsizes |grep -v 'chrM' > size.genome.no.mt
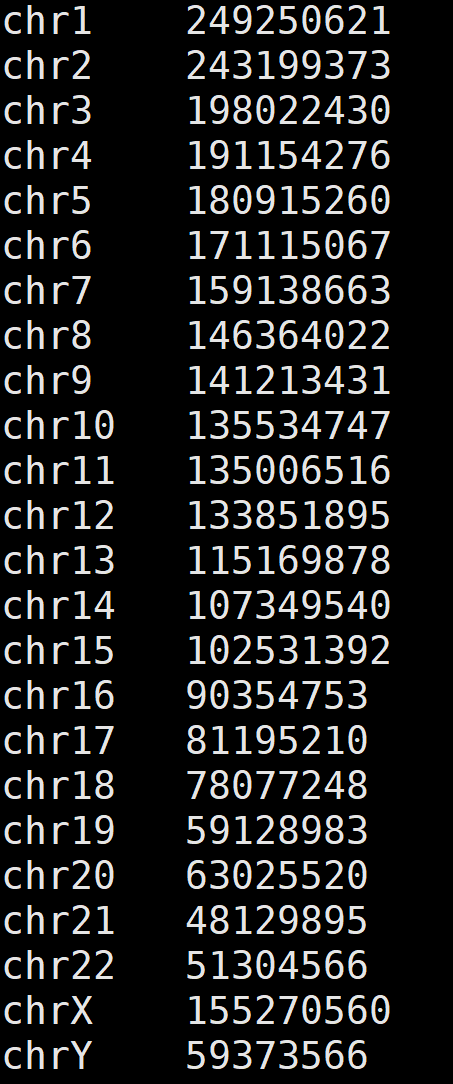
2. 窗口划分获取BIN文件
bedtools makewindows -g size.genome.no.mt -w 1000000 > windows.bed
- g sizes.genome是要划分的基因组,格式为两列:染色体、染色体长度
- w 1000000 为窗口大小为1M
- windows.bed为输出文件,格式为三列:染色体、区间开始位点、区间结束位点。
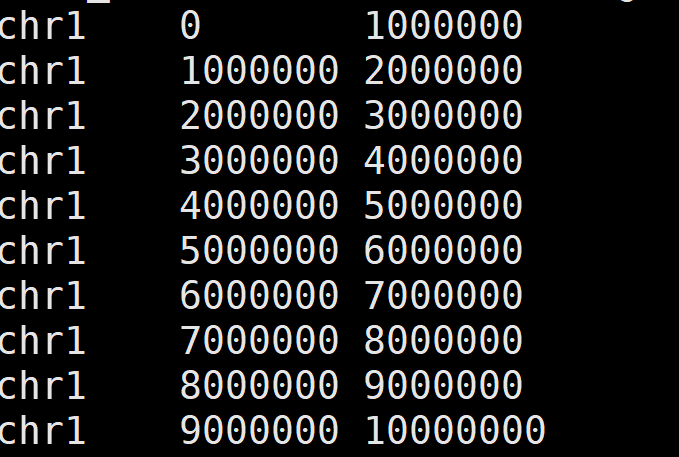
3. 常见统计 - 统计fasta序列每个窗口的GC含量
以获取参考基因组hg19.fa序列每1M窗口的GC含量为例
bedtools nuc -fi hg19.fa -bed windows.bed |cut -f 1-3,5 > gc.1M.bed
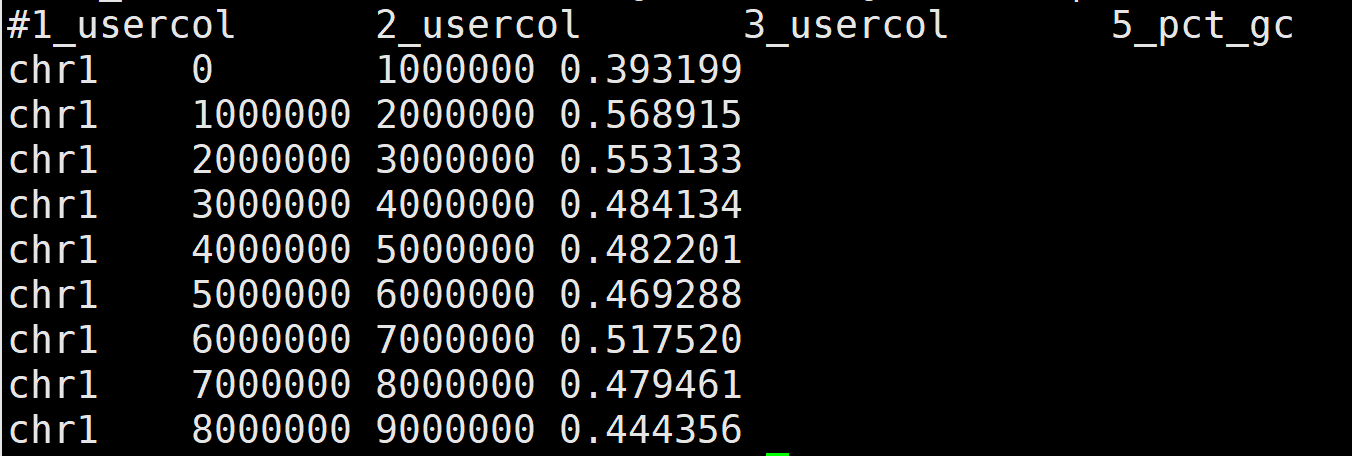
4. 常见统计 - 统计每个窗口的平均覆盖深度
bedtools coverage -a windows.bed -b Sample.sorted.bam > Sample.depth.txt
- Sample.sorted.bam 为比对后排序的bam文件
- windows.bed文件为步骤2获得的窗口划分BIN文件
- 生成的Sample.depth.txt文件共有7列,分别为序列编号、起始位置、结束位置、reads数、碱基数、区间大小、平均覆盖深度
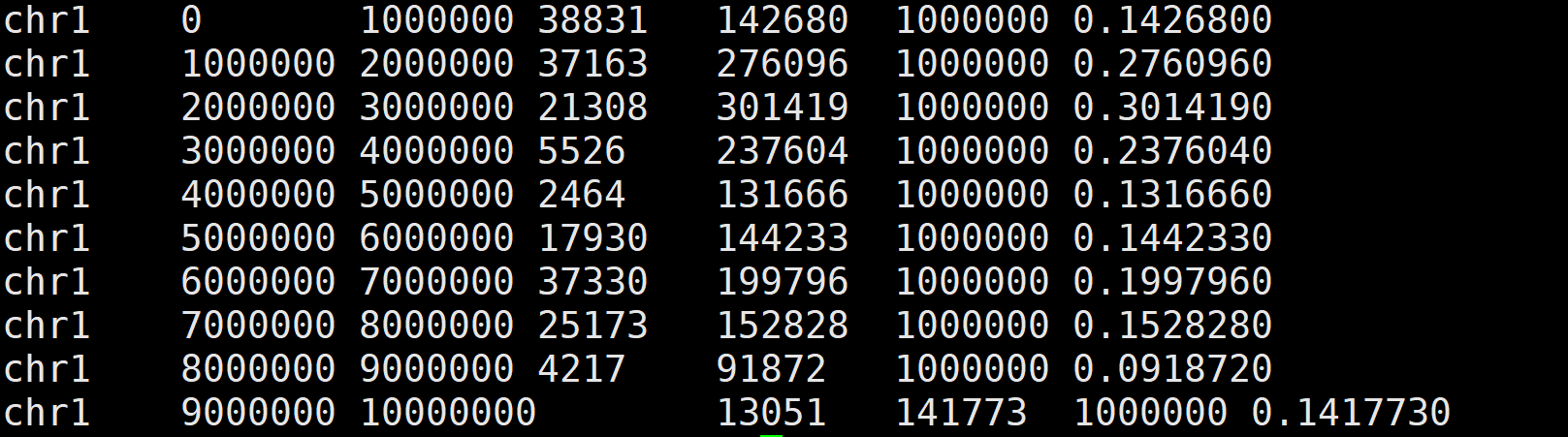
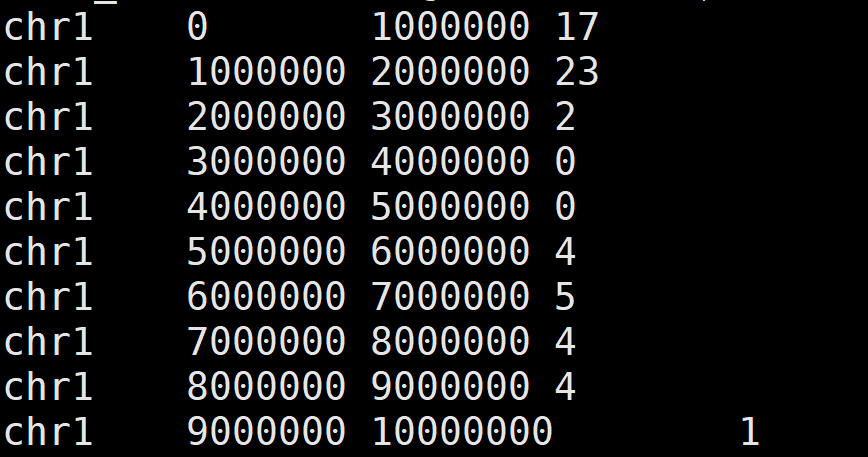
5. 常见统计 - 统计每个窗口的SNP变异位点数量
bedtools coverage -a windows.bed -b Sample.snp.filter.vcf -counts > snp.1M.txt
- windows.bed文件为步骤2获得的窗口划分BIN文件
- Sample.snp.filter.vcf为存储过滤后SNP变异位点的VCF文件
- snp.1M.txt 结果文件中共4列,分别为染色体、窗口起始位置、窗口结束位置和窗口SNP变异位点数量
生信软件文章推荐
生信软件1 - 测序下机文件比对结果可视化工具 visNano
生信软件3 - mapping比对bam文件质量评估工具 qualimap
生信软件4 - 拷贝数变异CNV分析软件 WisecondorX
生信软件7 - 多线程并行运行Linux效率工具Parallel
















 1593
1593











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











