







 文章介绍了如何使用WisecondorX进行拷贝数变异(CNV)分析,包括conda和pip安装方法,从比对文件(sam/bam/cram)转换成npz格式,建立参考,以及CNV变异检测。Zscore的设定用于过滤CNV,重要输出文件包括ID_aberrations.bed和ID_statistics.bed,同时软件提供可视化结果。
文章介绍了如何使用WisecondorX进行拷贝数变异(CNV)分析,包括conda和pip安装方法,从比对文件(sam/bam/cram)转换成npz格式,建立参考,以及CNV变异检测。Zscore的设定用于过滤CNV,重要输出文件包括ID_aberrations.bed和ID_statistics.bed,同时软件提供可视化结果。
 使用wisecondorX可进行拷贝数变异CNV的分析,作者在论文中对比了多种软件的使用效果,可自行根据自己的项目需要,判断是否使用。
使用wisecondorX可进行拷贝数变异CNV的分析,作者在论文中对比了多种软件的使用效果,可自行根据自己的项目需要,判断是否使用。
wisecondrX安装
# conda安装
conda install -f -c conda-forge -c bioconda wisecondorx
conda install bwa
# pip安装:
pip install -U git+https://github.com/CenterForMedicalGeneticsGhent/WisecondorX
使用wisecondrX做CNV分析
step1 将比对文件(sam/bam/cram)转换成npz格式
作者文章建议使用bowtie2进行比对,本人采用bwa进行的比对,并且对于比对后的bam文件不要进行过滤。
# bwa比对,以单端测序SE为例;双端测试可以行搜索
bwa mem -t 20 /public/reference/hg19.fasta sample_L01_F1.fq | samtools view -bSh - | samtools sort -@ 10 - -o ./sample.bam
# 将比对得到的bam文件转换为npz格式文件
WisecondorX convert sample.bam sample.npz
optional arguments:
--binsize BINSIZE Bin size (bp) (default: 5000.0)
step2 建立reference
至少需要50例健康人样本来建立reference,并且尽量保证男性和女性的比例为1:1,bin的长度可以设置不同的梯度,建议10kb~100kb之间。NIPT使用100kb bins进行评估,因为所有纳入NIPT畸变的宽度都比较大(至少5Mb)。
WisecondorX newref --cpus 20 --binsize 100000 control/*npz reference_100kb.npz
WisecondorX newref --cpus 20 --binsize 50000 control/*npz reference_50kb.npz
WisecondorX newref --cpus 20 --binsize 10000 control/*npz reference_10kb.npz
Create a new reference using healthy reference samples
optional arguments:
--yfrac YFRAC Use to manually set the y read fraction cutoff, which defines gender (default: None)
CNV变异检测
通过设置Zscore cutoff来过滤CNV,通常我会将Zscore设置为5。
WisecondorX predict --plot --bed sample.npz reference_100kb.npz
optional arguments:
--zscore ZSCORE z-score cut-off for aberration calling. (default: 5)
--gender {F,M} Force WisecondorX to analyze this case as a male (M) or a female (F) (default: None)
--add-plot-title Add the output name as plot title (default: False)
分析结果中比较重要的两个文件是ID_aberrations.bed和ID_statistics.bed。
ID_aberrations.bed文件中包含了拷贝数异常的CNV片段,格式如下:
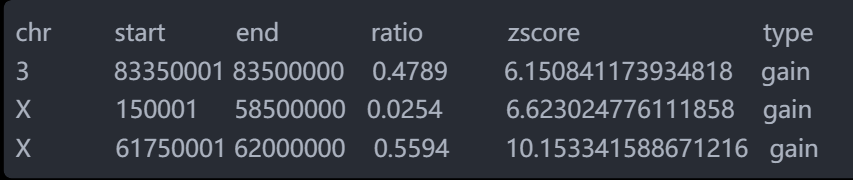 zscore列是Zscore值,一般在cutoff值附近的 是可疑点,需要重点关注。type列是表示gain重复或loss缺失。
zscore列是Zscore值,一般在cutoff值附近的 是可疑点,需要重点关注。type列是表示gain重复或loss缺失。
ID_statistics.bed文件中包含整条染色体的信息,包括ratio和Zscore,如果关注非整倍体变异的话可以仔细看一下这个文件。
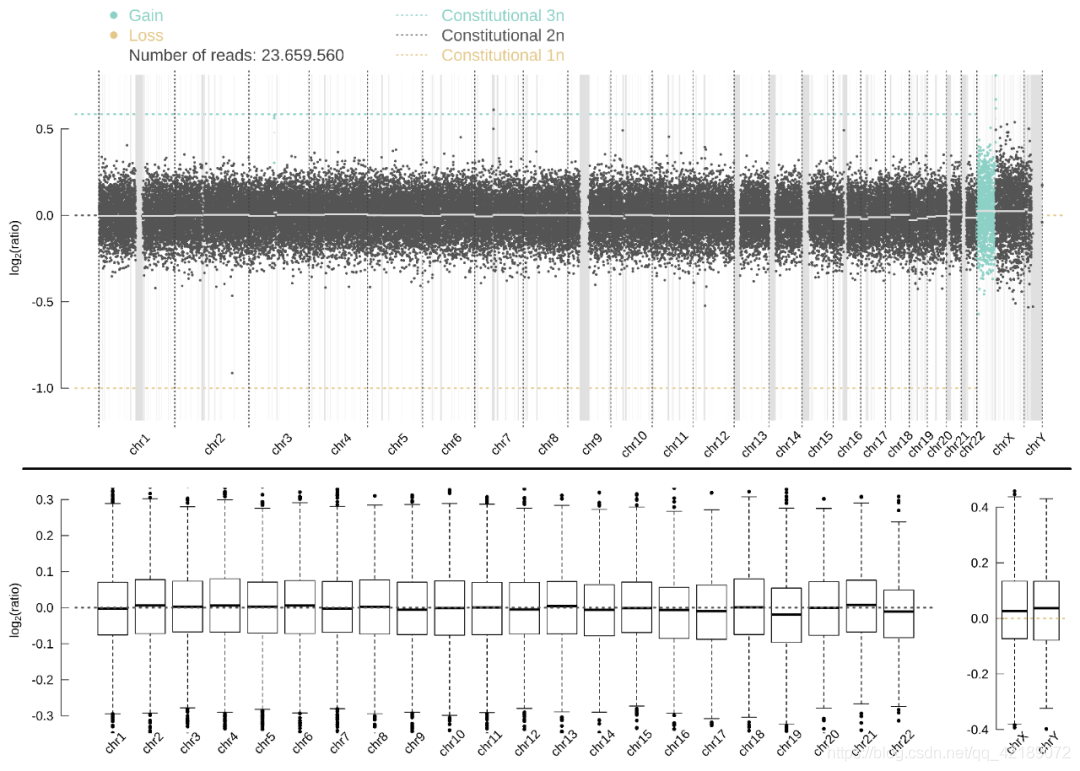
分析的时添加plot参数,WisecondorX还会生成可视化的结果,非常直观,包括全基因组和单条染色体的图形。



















 207
207

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











