







全基因组关联研究(GWAS)广泛应用于探究表型变异的遗传本质。通过利用全基因组范围内群体中高密度的分子标记,GWAS能够识别与复杂性状表型变异相关的分子标记,从而发掘与表型相关的基因,在作物改良育种方面,也发挥着非常重要的作用,通常基于基因组序列和高基因组覆盖的分子标记(例如单核苷酸多态性(SNP)标记)数据,使用GWAS分析手段用来分离农艺重要基因。
地球上高达70%的开花植物可能是多倍体,约40%-50%的栽培作物具有多倍体基因组,但到目前为止,已经发表的大量针对多倍体植物的GWAS调查中,常常使用单个亚基因组的同源基因进行基因分型和关联测试,而不是使用所有同源基因,这就导致无法在多倍体作物的全基因组水平上检测农艺重要同源位点。
近期,《Molecular Plant》在线发表了河南农业大学、中国农业科学院作物科学研究所与河南科技学院等多个单位的合作研究论文“Genome resources for the elite bread wheat cultivar Aikang 58 and mining of elite homeologous haplotypes for accelerating wheat improvement”。在该文献中,研究人员提出了一种新的优于GWAS的方法,即基于同源基因座的全基因组关联分析(HGWAS),可以对小麦多倍体作物的重要农艺性状进行检测分析。接下来,就和小编一起来看下这个新方法吧!

技术方法概述
研究材料:优良冬小麦品种矮抗58号(AK58)、中国春小麦(CS)、F2群体(AK58×CS)
技术手段:Illumina测序、PacBio单分子实时测序、10X Genomics、Hi-C技术、QTL分析、HGWAS分析、转录组测序
主要结果
1、AK58基因组概况
本研究基于Illumina测序、PacBio单分子实时测序、10X Genomics以及Hi-C技术组装得到了AK58染色体水平基因组,基因组大小为14.75Gb,将近97%的组装序列被锚定并按顺序排列在21个伪染色体上(图1)。
表1 AK58基因组组装概况
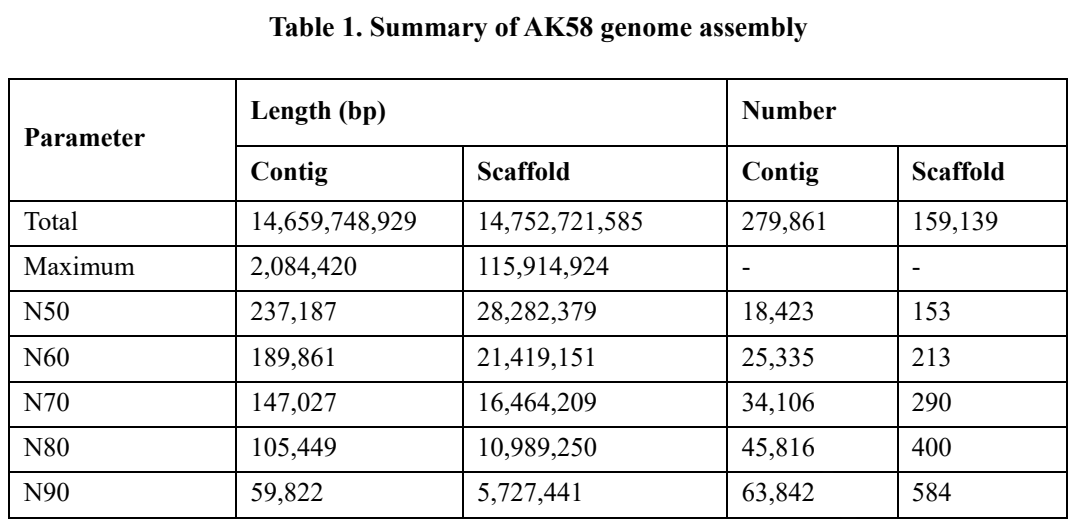
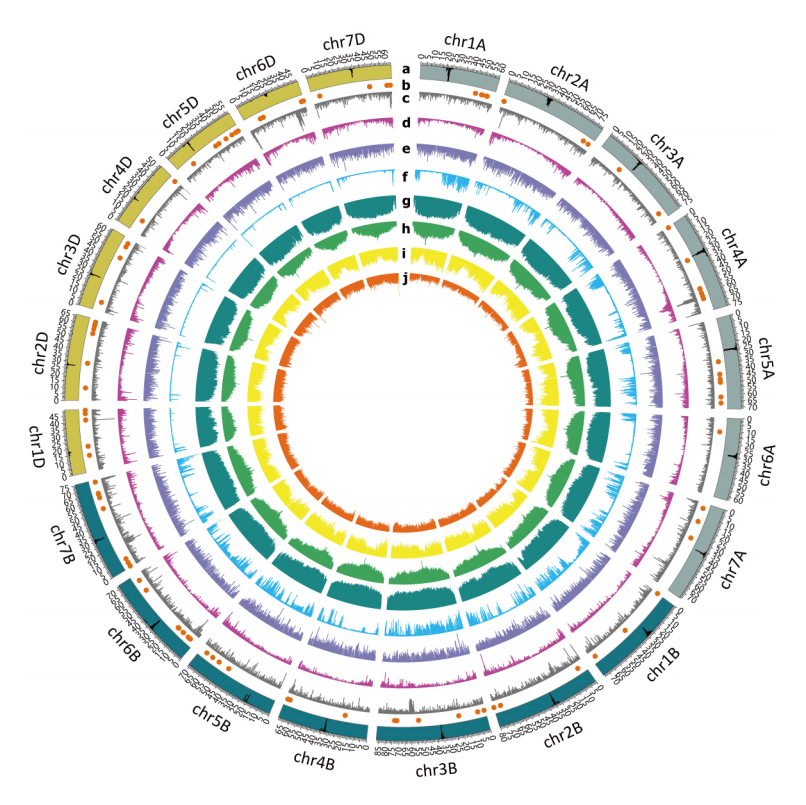
图1 AK58基因组
2、基因注释、多组学分析以及基因组综合数据库的构建
对AK58基因组进行了高置信度的蛋白编码基因(PCGs)的注释,总计119,448个PCGs。其中,亚基因组D相对于亚基因组B(38,538个)和亚基因组A(38,115个),具有更多的PCGs,达到了40,665个。
在AK58基因组中发现的TEs占基因组的85.3%,其中逆转录转座子和275个DNA转座子分别占基因组的67.09%和16.56%。在染色体的中间区域,转座子元件(TE)分布密集。这些区域的基因密度和重组率较低,如图1所示。在三个亚基因组中,反转录转座子的TE累积长度的排列顺序为B > A > D,而DNA转座子的TE累积长度的排列顺序为B > D > A。
本研究还开展了转录组、表观代谢组等研究,并基于基因、转座子(TE)、转录组、表观组、代谢组、突变体等信息构建了AK58基因组综合数据库(多组学数据库),为小麦及其它相关研究提供了丰富的多组学信息(图2)。
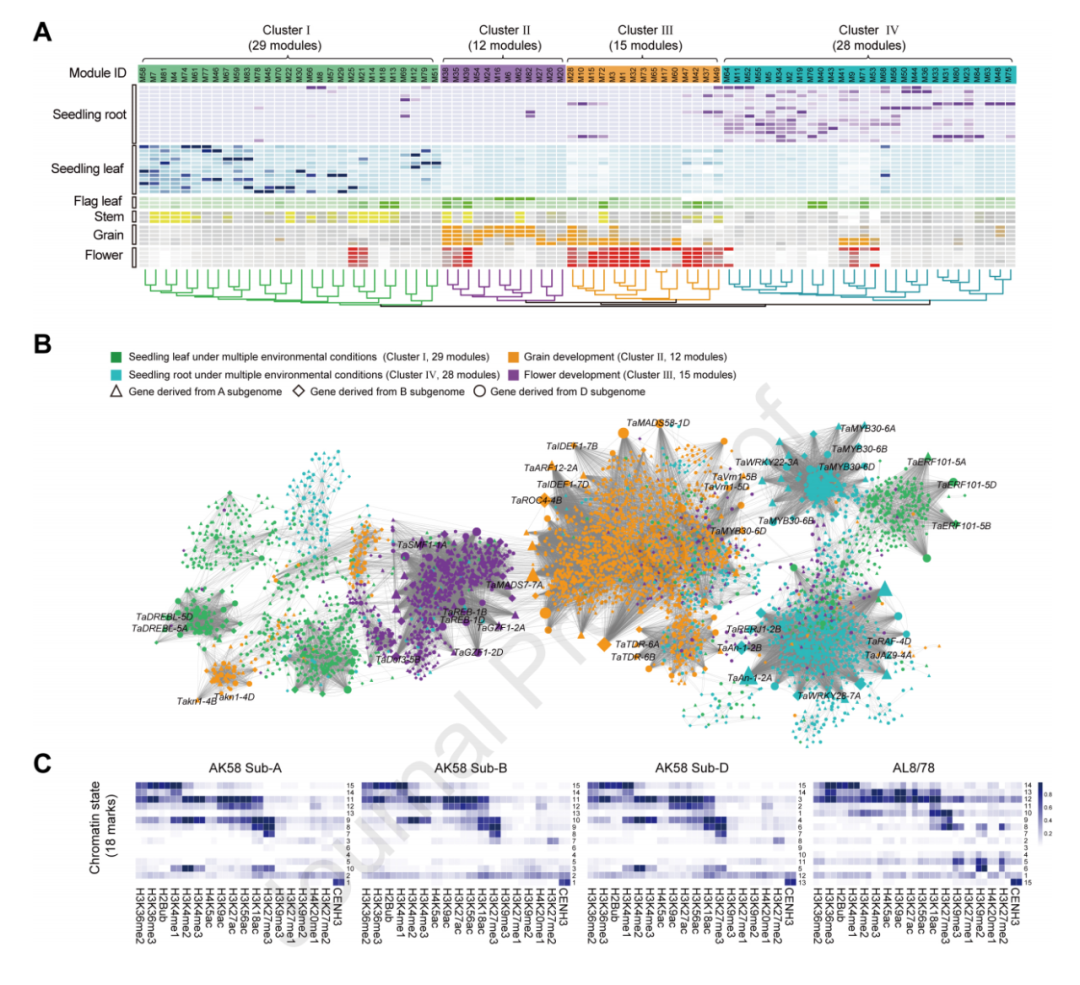
图2 多组学数据库
3、AK58和中国春(CS)的比较基因组分析
在AK58和CS的比较中,揭示了现代小麦改良中的基因组变化。共线性分析结果显示,约86%的AK58基因组和88%的CS基因组是共线的,其中最大的非同源区域位于Chr1B的短臂,这是因为AK58中存在1RS易位(图3B)。AK58和CS之间存在40,607,820个SNP和5,491,679个Indel变异。
通过比对AK58和CS基因组,本研究分析了基因型特异的结构变异(SV)。AK58特异的SV长度累积为183.3 Mb,占据了AK58基因组的1.3%,而CS特异的SV长度累积为107.8 Mb,占据了CS基因组的0.8%。GO术语富集分析显示,AK58和CS特异的SV基因在激酶活性和细胞蛋白修饰过程中富集,但是相比之下,AK58的SV基因更多地参与了光系统I和对创伤的响应。KEGG分析显示,AK58特异的SV基因在氧化磷酸化途径中显著富集,而CS特异的SV基因主要参与了植物与病原体的相互作用。
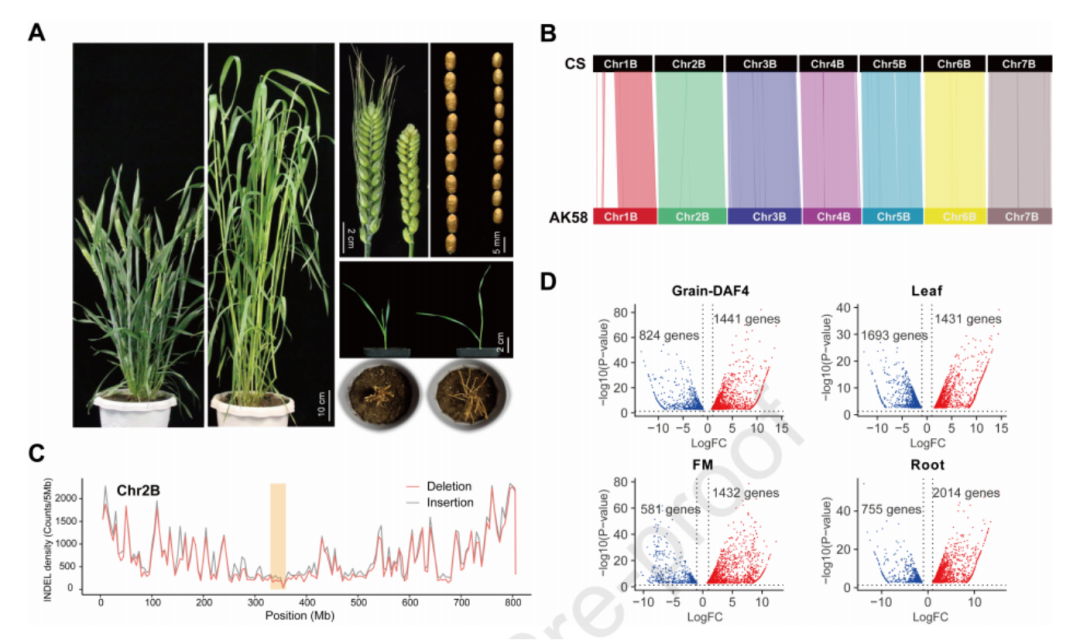
图3 AK58和中国春(CS)的比较基因组分析
4、基于同源基因座的全基因组关联分析(HGWAS)
通过将最接近每个同源基因的SNP标记定义为同源基因座的不同单倍型(图4A),本研究建立了同源基因座的标记-性状关联。利用HGWAS方法,研究人员发现了与叶片、穗长、籽粒和植株结构相关性状显著关联的393个基因座。其中,139个基因座在两个或更多环境中被检测到,并且对不同性状具有显著不同的遗传效应(图4B)。在这139个HGWAS基因座中,有123个基因座(占88.5%)每个都解释了超过10%的表型变异,因此被认为是主要控制性状的基因座。通过进一步比较HGWAS和传统GWAS的分析结果,研究人员发现HGWAS基因座解释的表型变异百分比通常高于其对应的GWAS基因座,具有更大的优势。
针对重要的HGWAS位点,研究人员对不同HHs之间的遗传效应进行了比较。在AK58和CS中都发现了多种优异的HHs。源自AK58的HHs反映了现代育种的选择效应,而存在于CS的HHs可能在现代育种选择中被遗弃,但仍具有潜在的利用价值。需要指出的是,研究人员发现了一些具有重要潜在价值、尚未被广泛利用的HHs,这对未来小麦改良具有重要意义。
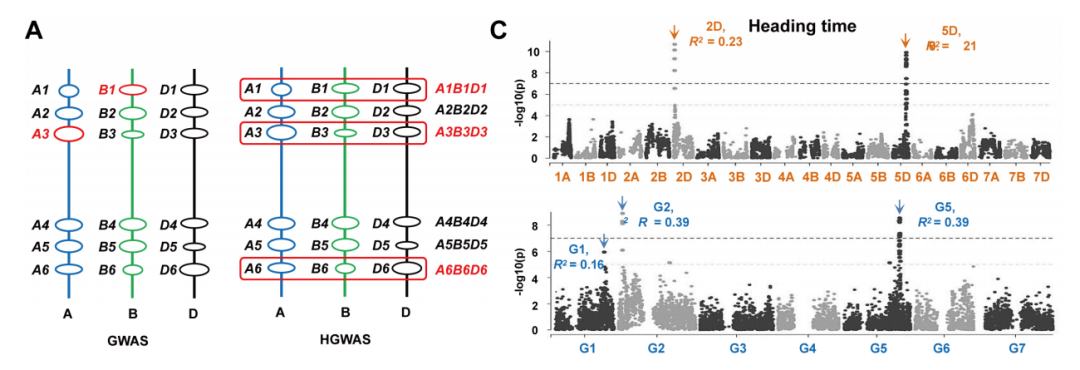
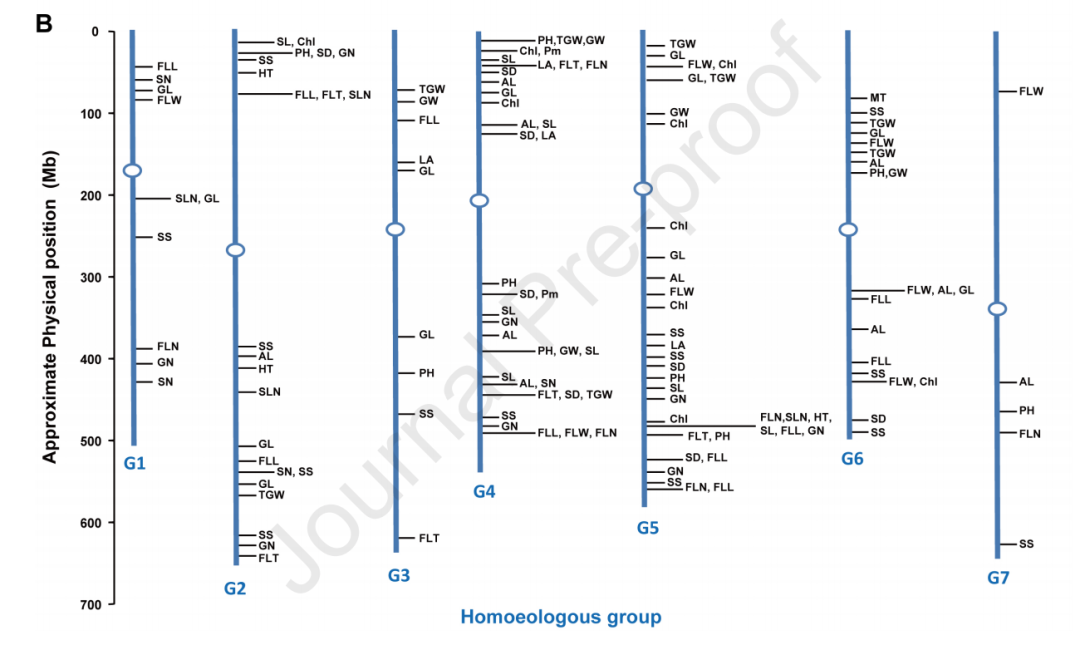
图4 HGWAS与传统GWAS的比较
总结
综上所述,本研究成功构建了AK58高质量染色体水平的基因组,并对其进行了多组学分析,形成了现代精英普通小麦品种矮抗58(AK58)的基因组资源。比较AK58与地方品种中国春(CS)揭示了小麦品种改良中发生的最新基因组变化。本研究还开发了基于同源位点的GWAS方法(HGWAS),并利用该方法鉴定了HGWAS位点。对于多倍体作物而言,HGWAS位点的鉴定,尤其是对优异HHs的挖掘与利用,将极大地丰富多倍体位点的遗传多样性,具有重要作用和利用价值。这对未来进一步解析和利用多倍体优势、培育更多更好的新品种具有重要意义。
参考文献
Genome resources for the elite bread wheat cultivar Aikang 58 and mining of elite homeologous haplotypes for accelerating wheat improvement. Molecular Plant, 2023.
















 1710
1710











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











