






技术背景
分子动力学模拟(Molecule Dynamics Simulation,MD),本质上是一门采样技术。通过配置力场参数、拓扑结构和积分器,对一个给定的体系不断的采样,最终得到一系列的轨迹。那么得到分子动力学模拟的轨迹之后,如何使用后分析工具进行轨迹分析,也是一项很重要的工作。目前来说,基于Python的开源工具MDAnalysis(简称mda)是一个比较常用的MD后分析工具。本文主要介绍基于MindSponge分子动力学模拟框架生成了相应的轨迹之后,如何使用MDAnalysis工具进行分析。
环境配置
需要说明的是,MindSponge当前主要有两个版本,一个是华为MindSpore下的官方仓库MindScience,这里面包含了多个工具的正式发布版本,其中也有MindSponge,相对而言功能比较稳定,但是需要编译构建和安装使用。另外一个仓库是MindSponge,是MindSponge开发团队维护的一个develop版本,这个仓库只要git clone下来就可以测试和使用。本文章中的相关代码是基于后者来实现的,暂时没上正式版仓库。关于MindSponge的安装和基本使用方法,可以参考下之前的文章,所有的内容都是开源免费的。
然后MDAnalysis可以用pip直接安装(这里我们使用的是pip清华源):
$ python3 -m pip install mdanalysis --upgrade
Looking in indexes: https://pypi.tuna.tsinghua.edu.cn/simple
Requirement already satisfied: mdanalysis in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (2.7.0)
Requirement already satisfied: numpy<2.0,>=1.22.3 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (1.24.0)
Requirement already satisfied: GridDataFormats>=0.4.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (1.0.2)
Requirement already satisfied: mmtf-python>=1.0.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (1.1.3)
Requirement already satisfied: joblib>=0.12 in /home/dechin/.local/lib/python3.9/site-packages (from mdanalysis) (1.2.0)
Requirement already satisfied: scipy>=1.5.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (1.10.1)
Requirement already satisfied: matplotlib>=1.5.1 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (3.7.1)
Requirement already satisfied: tqdm>=4.43.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (4.65.0)
Requirement already satisfied: threadpoolctl in /home/dechin/.local/lib/python3.9/site-packages (from mdanalysis) (3.1.0)
Requirement already satisfied: packaging in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (23.0)
Requirement already satisfied: fasteners in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (0.19)
Requirement already satisfied: mda-xdrlib in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mdanalysis) (0.2.0)
Requirement already satisfied: mrcfile in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from GridDataFormats>=0.4.0->mdanalysis) (1.5.0)
Requirement already satisfied: contourpy>=1.0.1 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (1.0.7)
Requirement already satisfied: cycler>=0.10 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (0.11.0)
Requirement already satisfied: fonttools>=4.22.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (4.38.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (1.4.4)
Requirement already satisfied: pillow>=6.2.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (9.4.0)
Requirement already satisfied: pyparsing>=2.3.1 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (3.0.9)
Requirement already satisfied: python-dateutil>=2.7 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (2.8.2)
Requirement already satisfied: importlib-resources>=3.2.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from matplotlib>=1.5.1->mdanalysis) (5.12.0)
Requirement already satisfied: msgpack>=1.0.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from mmtf-python>=1.0.0->mdanalysis) (1.0.5)
Requirement already satisfied: zipp>=3.1.0 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from importlib-resources>=3.2.0->matplotlib>=1.5.1->mdanalysis) (3.11.0)
Requirement already satisfied: six>=1.5 in /home/dechin/anaconda3/envs/mindsponge/lib/python3.9/site-packages (from python-dateutil>=2.7->matplotlib>=1.5.1->mdanalysis) (1.16.0)安装完成后,就可以先用MindSponge生成一个用于后分析的轨迹,再调用MDAnalysis进行分析。
生成轨迹
这里我们使用的案例轨迹,还是前一篇文章中所用到的能量极小化的一个案例。模拟的分子是这个样子的:

分子动力学模拟的相关代码如下:
from mindspore import nn, context
import numpy as np
import sys
# 添加sponge所在的路径,这样就不需要安装即可直接使用
sys.path.insert(0, '../..')
from sponge import ForceField, Sponge, set_global_units, Protein
from sponge.callback import RunInfo, WriteH5MD, SaveLastPdb
from sponge.colvar import Distance, Angle, Torsion
# 配置MindSpore的执行环境
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
# 配置全局单位
set_global_units('A', 'kcal/mol')
# 定义一个基于case1.pdb的分子系统
system = Protein('../pdb/case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
# 定义一个amber.ff99sb的力场
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定义一个学习率为1e-03的Adam优化器
min_opt = nn.Adam(system.trainable_params(), 1e-03)
cv_bond = Distance([0, 1])
cv_angle = Angle([0, 1, 2])
cv_dihedral = Torsion([0, 1, 2, 3])
# 定义一个用于执行分子模拟的Sponge实例
md = Sponge(system, potential=energy, optimizer=min_opt, metrics={'bond': cv_bond, 'angle': cv_angle,
'dihedral': cv_dihedral})
# RunInfo这个回调函数可以在屏幕上根据指定频次输出能量参数
run_info = RunInfo(20)
# WriteH5MD回调函数,可以将轨迹、能量、力和速度等参数保留到一个hdf5文件中,文件后缀为h5md
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False, save_last_pdb='last_pdb.pdb')
# 保存PDB文件
bonds = np.array([[0, 56], [6, 10]], np.int32)
# 开始执行分子动力学模拟,运行2000次迭代
md.run(200, callbacks=[run_info, cb_h5md])运行结束后,会在当前路径下生成一个名为last_pdb.pdb的构象文件和一个test.h5md的轨迹文件。关于h5md格式的轨迹文件,可以用silx这个工具来进行直观的可视化:

这是体系能量极小化过程中的能量变化曲线:
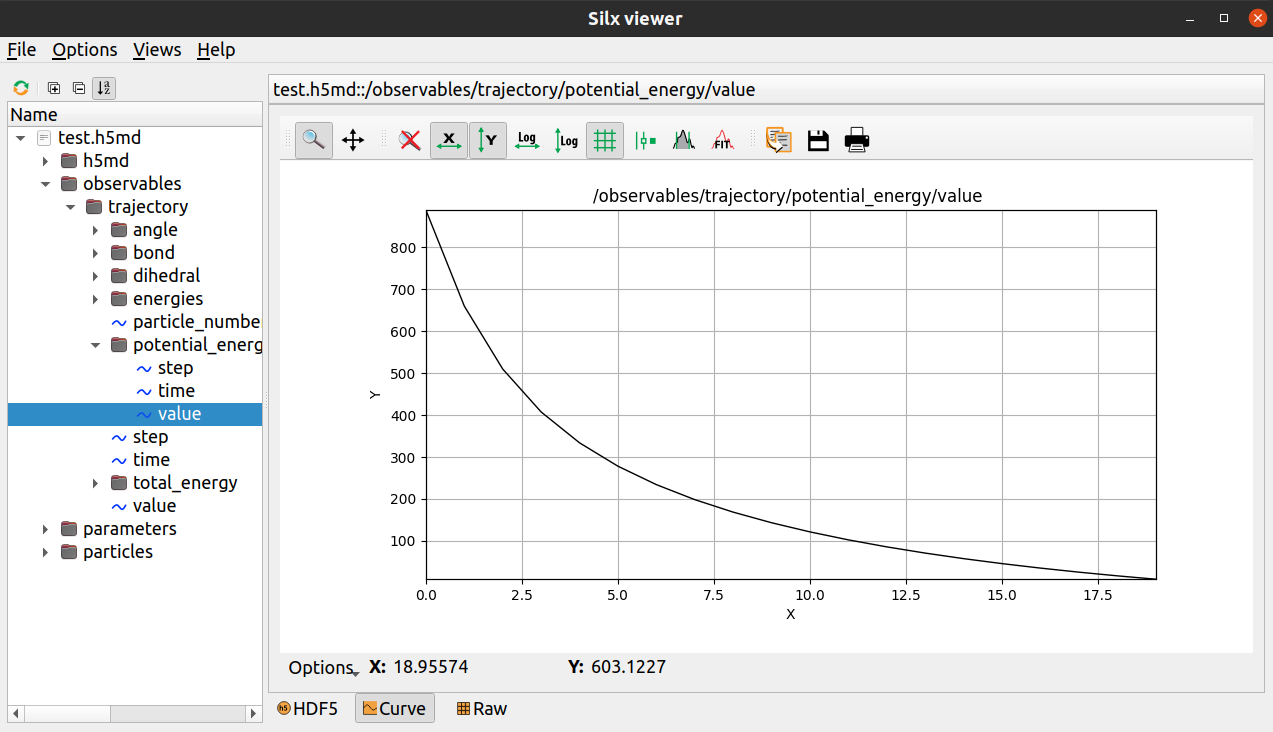
并且保存了轨迹数据:

MDAnalysis分析
使用MDAnalysis进行分析的主要流程,就是用拓扑结构文件和轨迹文件构建两个MDAnalysis.Universe对象。这里拓扑结构文件可以使用pdb文件,但要求pdb文件中包含有CONECT成键相互关系,否则跟成键相互作用相关的内容使用mda无法分析,MindSponge所生成的pdb文件中是包含了成键关系信息的。再者就是h5md也是mda所支持的轨迹文件扩展名,使用MindSponge生成的轨迹可以直接用mda加载:
import MDAnalysis as mda
u = mda.Universe('last_pdb.pdb', 'test.h5md')加载完之后,我们可以打印其中的一些关键信息,比如原子类型和残基类型等:
print('Atom Types List:\n', u.atoms)
# Atom Types List:
# <AtomGroup [<Atom 1: N of type N of resname ALA, resid 1 and segid A and altLoc >, <Atom 2: CA of type C of resname ALA, resid 1 and segid A and altLoc >, <Atom 3: CB of type C of resname ALA, resid 1 and segid A and altLoc >, ..., <Atom 55: HB1 of type H of resname ALA, resid 4 and segid A and altLoc >, <Atom 56: HB2 of type H of resname ALA, resid 4 and segid A and altLoc >, <Atom 57: HB3 of type H of resname ALA, resid 4 and segid A and altLoc >]>
print('Residue Types List:\n', u.residues)
# Residue Types List:
# <ResidueGroup [<Residue ALA, 1>, <Residue ARG, 2>, <Residue ALA, 3>, <Residue ALA, 4>]>
print('Step 0 Coordinates Shape:\n', np.array(u.coord).shape)
# Step 0 Coordinates Shape:
# (57, 3)
print('C Atoms:\n', u.select_atoms('name C'))
# C Atoms:
# <AtomGroup [<Atom 4: C of type C of resname ALA, resid 1 and segid A and altLoc >, <Atom 22: C of type C of resname ARG, resid 2 and segid A and altLoc >, <Atom 40: C of type C of resname ALA, resid 3 and segid A and altLoc >, <Atom 50: C of type C of resname ALA, resid 4 and segid A and altLoc >]>
print('Contact Map:\n', contact_matrix(np.array(u.coord)))
# Contact Map:
# [[ True True True ... True True True]
# [ True True True ... True True True]
# [ True True True ... True True True]
# ...
# [ True True True ... True True True]
# [ True True True ... True True True]
# [ True True True ... True True True]]
print(u.bonds)
# <TopologyGroup containing 56 bonds>然后是一些跟轨迹相关的条目:
print('Number of Frames in Trajectory:\n', u.trajectory.n_frames)
# Number of Frames in Trajectory:
# 20
print('Number of Atoms:\n', u.trajectory.n_atoms)
# Number of Atoms:
# 57
print(u.trajectory.has_positions)
# True
print(u.trajectory.ts.positions.shape)
# (57, 3)
print(u.trajectory.ts.has_velocities)
# False
print(u.trajectory.ts.has_forces)
# False
print(u.trajectory[0].data)
# {'trajectory': 10, 'time': 0.010000000707805157, 'step': 10}
print(u.trajectory[1].positions[0])
# [ -0.11355944 -11.455442 -0.79421705]因为我们在定义CallBack的时候没有在轨迹中保存速度参量和力参量,因此这里has_velocities和has_forces两个的值都是False,但实际上我们是可以支持在中间轨迹把这两个参量写入到h5md文件中的。由于轨迹有很多帧,在mda里面我们可以直接对u.trajectory使用索引,来定位到特定的某一帧,再导出自己所需要的参量。除了单点分析,我们还可以定义一个reference trajectory来计算RMSD等参数:
ref = mda.Universe('last_pdb.pdb', 'test.h5md')
R = RMSD(u, ref, select="backbone", groupselections=['backbone and resid 1-4'])
R.run()
rmsd = R.results.rmsd.T
print (rmsd[2])更多的MDAnalysis工具的使用方法和函数接口,可以参考MDAnalysis官方文档或者是这个中文翻译版文档。
总结概要
这篇文章我们主要介绍了MindSponge分子动力学模拟软件如何跟后分析工具MDAnalysis相配合的方法,其主要操作流程就是调用MindSponge自带的CallBack来输出拓扑文件和轨迹文件给MDAnalysis,然后就可以调用MDAnalysis的相关分析函数接口,十分的方便。
版权声明
本文首发链接为:https://www.cnblogs.com/dechinphy/p/mda-mds.html
作者ID:DechinPhy
更多原著文章:https://www.cnblogs.com/dechinphy/
请博主喝咖啡:https://www.cnblogs.com/dechinphy/gallery/image/379634.html















 2035
2035











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









