







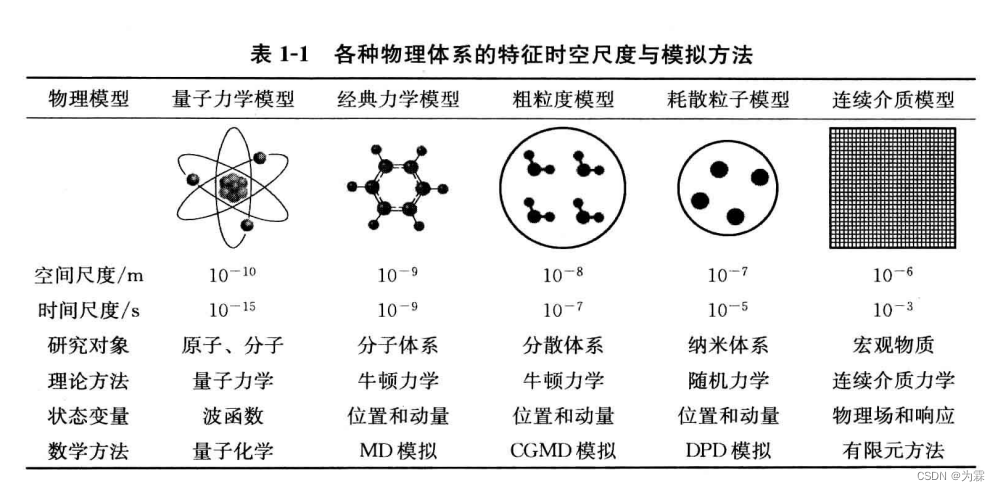
提纲



第一章 绪论
1.1 分子动力学模拟的发展历史
- 定义:研究分子或者分子体系结构与性质的计算方法
- 研究基础:经典力学
- 分子力学方法(MM):优化单个分子总能量得到稳定构型
- 蒙特卡洛(MC):通过反复采样分子体系位形空间计算其总能量
- 分子动力学(MD):数值求解分子体系运动方程得到体系相轨迹,统计体系的结构特征与性质
- 发展历史—研究模型由单原子转向多原子,是否考虑化学键约束,力场变化,数值求解方法发展,温度与压力调控
- 刚性小球完全碰撞
- 具有lennard-jones势函数的Ar原子体系→分子运动方程数值方法
- 单原子分子体系
- 多原子分子体系
- 如果化学键伸缩势、键角弯曲势都由势函数描述——求解方法与单原子分子体系相同
- 化学键约束下不具备内部自由度的多原子分子——近似为刚体
- 具有内部自由度的分子 - 温度与压力调控与统计系综的实现
- NVE
- NVT——变标度&恒温扩展法
- 差分误差积累——温度与压力调控方法、差分算法纳入统一的理论框架
- 分子力场发展
- AIMD模拟:基于量子化学方法直接计算所有分子内和分子间的相互作用,不需要输入经验的力场模型
- 力的计算与节省时间——势函数的截断与修正,近邻列表算法,格子索引
1.2 发展趋势
- 计算技术:并行加速比提升,GPU使用与编程
- MD算法与分子模型发展方向
- 算法改进:减少最耗时部分——相互作用力计算
- 简化模拟体系描述:限制内部自由度或者延长模拟积分步长
- 多尺度与介观体系MD模拟:利用粗粒度模型把分子力场模型和连续介质模型有机结合

- AIMD模拟与反应性分子力场:反应性力场不能计算原子间相互作用,但允许化学键的断裂和生成;包括Reaxff MD和MS-EVB
- 多尺度与介观体系MD模拟:利用粗粒度模型把分子力场模型和连续介质模型有机结合
第二章 分子的物理模型
模型:对客观世界近似的方程、公式
2.1 在化学中的应用
- 几何模型:分子内不同原子及原子间距离的相对尺度
- 振动模型:为了理解红外光谱,引入化学键伸缩和键角弯曲的谐振子模型或其他振动模型
- 力场模型:几何模型基础上引入分子内和分子间相互作用及其能量的概念
- 量子力学:为了理解电子能谱,基于薛定谔方程或密度泛函研究电子的运动及规律
2.2 几何模型
- Dalton提出原子模型:具有固定体积、质量(后修正为核电荷)的球体
- Avogadro 提出相同体积具有相同的分子数量,其质量由相对分子质量决定,得到了12gC12中原子数量
- 实验证实:2D NMR;3D NMR;XRD
2.3 经典力学模型——引入分子间和分子内相互作用的概念,将分子模型和能量联系起来
2.3.1 共价键的伸缩运动
-
来源:相同结构分子具有不同的红外光谱
-
共价键的谐振子模型(频率和势能计算函数)
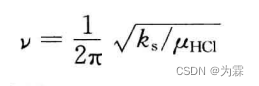
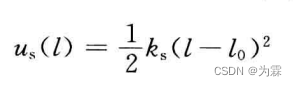
-
理想谐振子模型作为一级近似,三次及以上次幂的Taylor作为近似,则共价键势函数近似为:
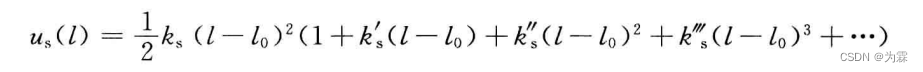
-
远离参考键长,呈现Morse势函数
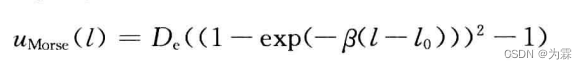
-
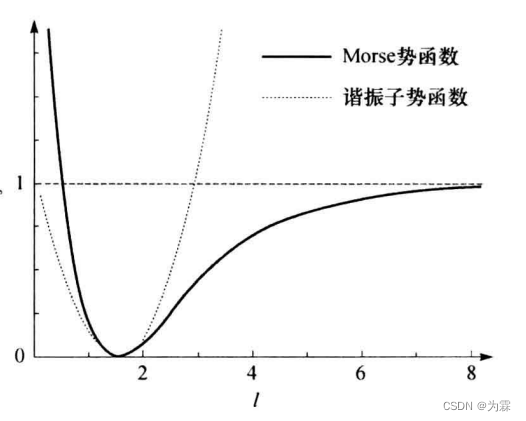
Morse势函数在远离键长时更准确,谐振子模型不允许键的断裂,但两者在参考键长附近计算结果类似
2.3.3 键角的弯曲振动
- H2O分子内振动模式包括:H-O对称伸缩振动,H-O不对称伸缩振动,键角弯曲振动
- 一般N个原子组成的分子,最少有N-1个化学键,一般有2(N-1)+N-3个内部振动模式
- 键角偏离参考值时的势函数:
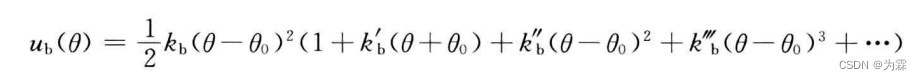
2.3.4 二面角扭曲运动
- 相较于键的伸缩和键角弯曲
- 达到平衡状态速度更慢
- 对分子构型起决定作用
- 扭曲势能较弱:在1~10kcal/mol左右,受分子热运动和周边分子原子位阻严重影响
- 对总体能量贡献小,但重要性高
- 和非键像话作用处在相同强度范围
- 下列公式近似:
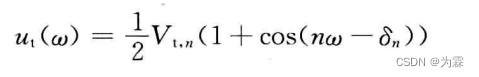
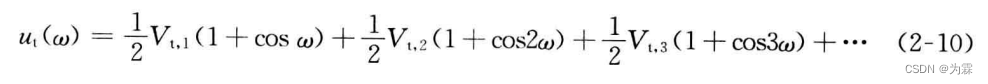
2.3.4 离面弯曲运动
对于SP2杂化碳原子的分子,必须保持其平面构型,目前,常用离面弯曲运动、赝扭曲运动和翻转运动描述结构对平面构型的偏离。如:
-
离面扭曲势能
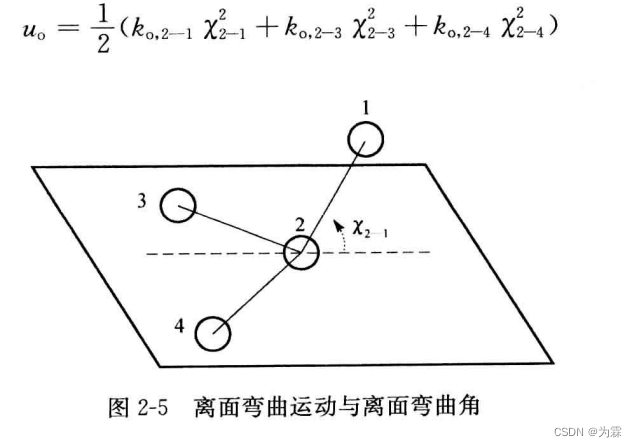
-
赝扭曲势能
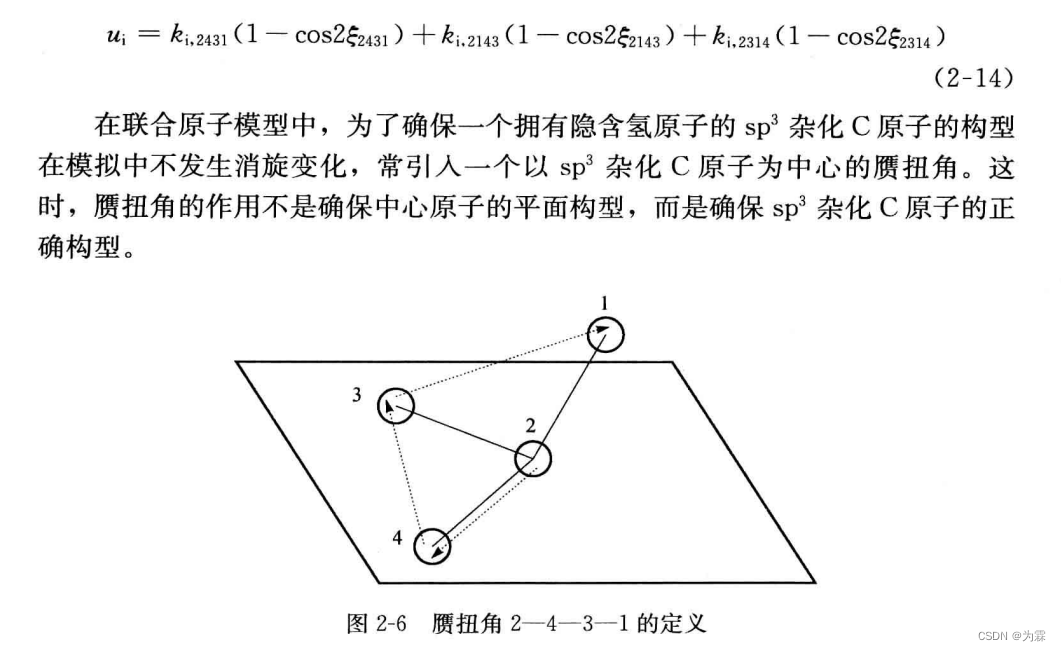
赝扭曲不是为了保证中心原子的平面构型,目的是为了保证SP3的碳原子杂化构型 -
翻转运动
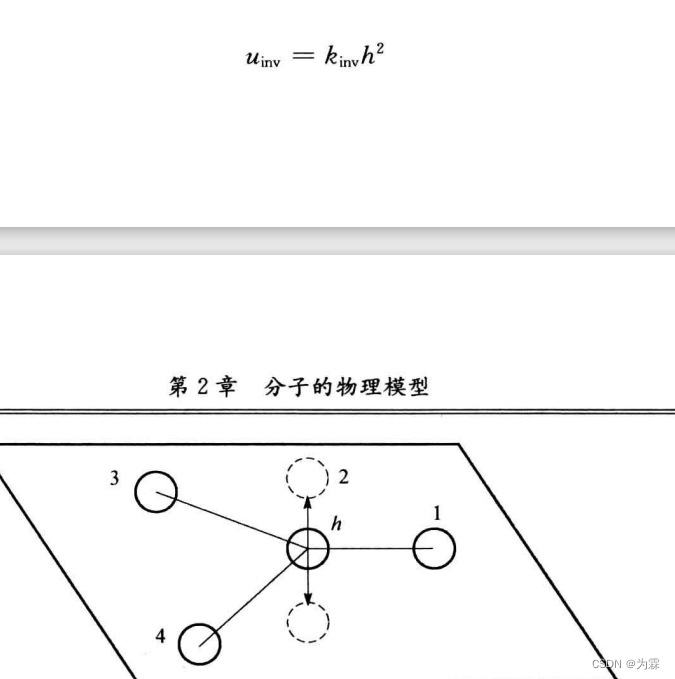
2.3.6 交叉项
- 键角和键角的两个化学键之间的耦合势能
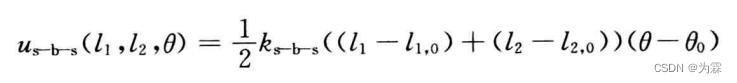
- 伸缩和伸缩,伸缩和扭曲,弯曲-弯曲,弯曲-扭曲耦合的势函数:
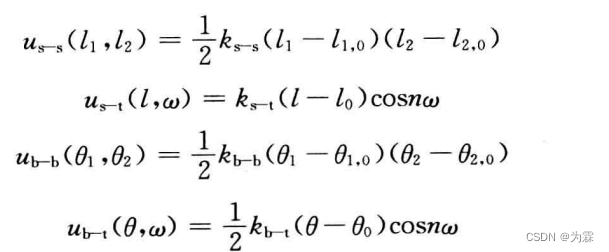
2.3.8 分子内的非键相互作用
相距超过三个化学键的两个原子中间,如果缺少非键相互作用就容易在空间上重叠。非键相互作用包括库伦相互作用、范德华作用和氢键
2.3.9 分子总势能的静电力场展开

注意:量子力学中,化学键的键能不与任何一个算符对应,不能通过薛定谔方程直接得到,把分子总能量分解为以上的部分具有很大的随意性。
2.4 分子的量子力学模型
电子不遵循静电力学,但遵循量子力学,基本方程是薛定谔方程
定态薛定谔方程:

第三章 分子间相互作用
3.1 分子间相互作用与势函数
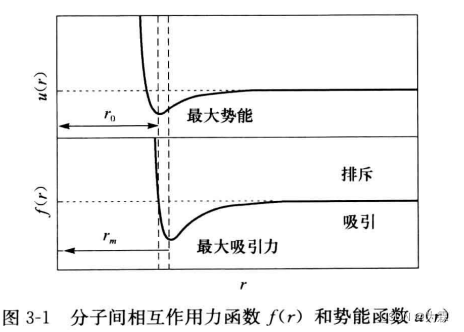
- 球形对称单原子分子间的相互作用力,只与原子核之间的距离有关,力和势函数和r之间的关系如下:
- 势函数和分子间作用力关系如下:
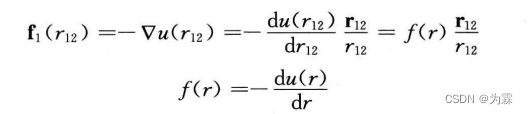
- 势函数决定了物质的性质,可以说,正是由于复杂多样的分子间相互作用势函数,决定了胶体,高分子,生物分子及超分子体系等复杂多样的性质
3.2 分子间特殊势函数
3.2.1 硬球势
- 势函数非常复杂,势函数和物质性质之间的关系也非常复杂,难以根据数函数用理论方法得到分子性质
- 简单的分子体系,如硬球势之间的简单假想分子体系,才能通过统计力学方法求得系列virial系数Bn,以及热力学性质解析解
- MD模拟得到的性质和统计力学求得的精确理论结果对比,改进MD模型
- 实际上,MD得到的分子体系性质,受分子模型可靠性和MD模拟方法可靠性双重英系那个
- 硬球势——最简单的势函数,把分子近似为直径为r,相互之间没有吸引力的硬球
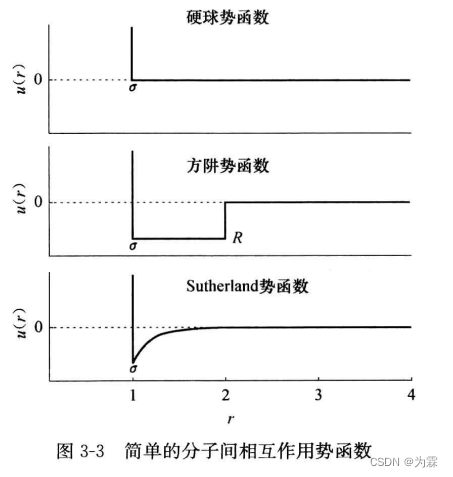
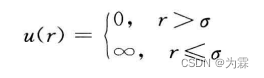
- 硬球势特点:分子质心距离大于r,相互作用力为0,相互作用质到质心间距离等于r,完全弹性碰撞而远离
3.2.2 方阱势函数
硬球势之间没有吸引力作用,球体不能凝结为液体或者固体,因此必须引入吸引力。最简单的吸引作用的势函数是方阱势。
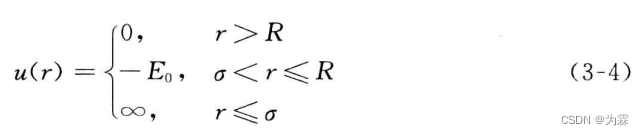
与硬球势不同的是增加了一个势井,厚度为R-r,深度为E0
方阱势能函数特点:(1)不连续(2)距离>R,无相互作用力(3)距离<R,收到巨大的吸引力,体系势能降到-E(4)分子距离小于r,分子发生弹性碰撞不能继续靠近
3.2.3 Sutherland势函数
- 解决方阱势不连续的问题,引入连续吸引力的势函数
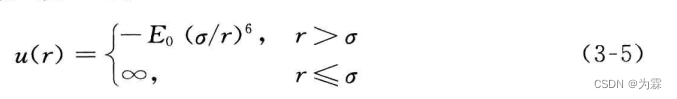
- 特点(1)吸引力连续,排斥力不连续(2)MD需要连续的势函数一阶导数,计算其分子间相互作用力;连续的二阶导数,进行分子结构优化,否则容易不稳定
3.3 分子间相互作用的起源——物理相互作用
四种相互作用力:万有引力,强核力,弱核力,电磁力。强、弱核力是短程力,主要存在原子核尺度内,万有引力极弱,因此分子间作用力是某种形式的电磁相互作用
分子间作用力:(1)物理相互作用,静电相互作用及其诱导的色散作用(2)弱化学作用:电子转移引起的化学作用,氢键作用和缔合作用
3.3.1 库伦势
- 来源:中性分子的残余电荷和离子间的相互作用
- 表达式:
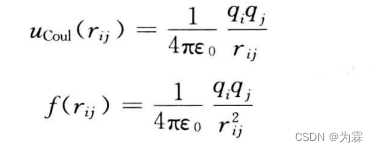
- 特点:和距离的一次方成反比,是长程相互作用力,不能使用计算近程相互作用时常用的截断近似
- 计算方法:Ewald求和和反应场算法
3.3.2 点电荷和偶极子相互作用势
- 偶极子:正负电荷中心不重合
- 偶极矩:负电荷中心指向正电荷中心的矢量
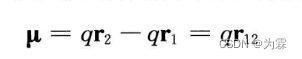
- 偶极矩单位:Debye,代表1A的带1 e.s.u和-1.e.s.u.的点电荷形成的偶极矩,1D=2.3354*10E-30 C·m
- 多原子分子的偶极矩定义:

- 一个点电荷q和偶极矩之间的相互作用势(适用于点电荷和偶极子中心之间的距离远大于偶极子尺寸)
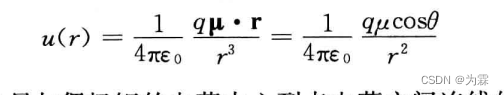
- 对不同空间取向取热力学平均以后得到:
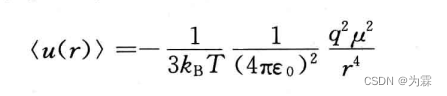
3.3.3 偶极子和偶极子之间的相互作用势能
两个相距很远,分别位于r1和r2上的偶极子u1和u2之间的相互作用势能可以用下式表达:
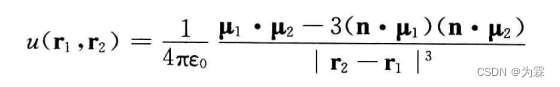
其中,n为r12方向上的鹅单位矢量,因此上式可以写成
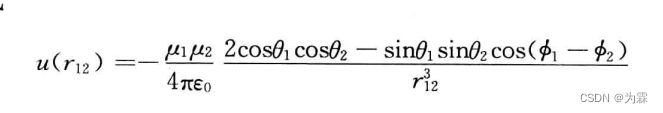
同样,对偶极子与偶极子取热力学平均得到:
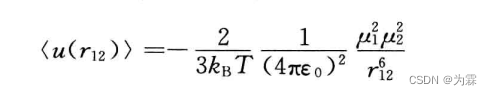
3.3.4 四极子
为了定义CO2这种偶极矩为零的分子间相互作用力,定义了四极子,八级子,十六极子概念
对不同空间取向取平均以后,点电荷与四极子,偶极子与四极子,四极子与四极子之间的相互作用势分别是
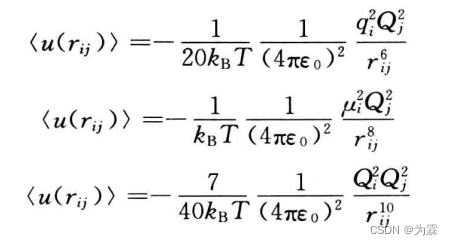
3.3.5 诱导作用
- **定义:**虽然非极性分子正负电荷中心重合,但是在外电场作用下电子云会沿着电场方向重排,引起正负电荷中心的重排。诱导偶极矩与外电场E成正比,结果如下:

- 极化率还可以用一个3X3的极化体积张量体积表达,因为分子体积越大,电子越多,越容易被极化
- 带电量为q的粒子与非极性分子之间的诱导能分两个部分

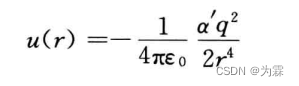
- 一般来说,偶极矩,四级距其诱导作用的贡献较小,实际应用中往往把各种高价矩之间的诱导能,作为有效能概括到rE-6项目当中
3.3.6 色散作用
- 根据paoli不相容原理,He,Ne,Ar等电子球形分布的稀有气体之间只有排斥力,无法形成液体,事实上还是可能凝结为液体。这是因为色散作用
- 来源: 瞬时偶极矩和诱导偶极矩之间的相互作用。色散作用是一种相互作用力,可以使体系能量变低,两个分子之间的色散能可以写成
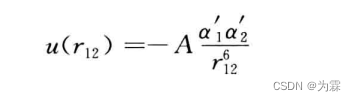
3.3.7 分子间的排斥作用
分子间排斥作用对体系势函数的贡献与分子间的距离呈指数关系:

3.4 氢键相互作用
- 分子间弱键作用力,D-H···A,D是供体体,A是受体,一般是N,O,F等体积小电负性高的分子。A必须有孤对电子,可以向H转移电子。
- 氢键作用势函数:

- 更复杂的势函数可以引入氢键的键角D-H···A依赖关系,如YETI势函数

3.5 常用分子间相互作用势函数
3.5.1 Lenard-Jones 势函数
- 第一种形式:
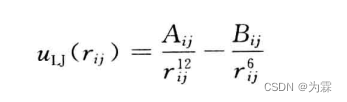
对应的作用力函数:
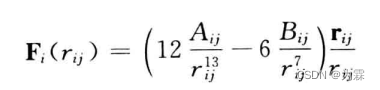
- 第二种形式:
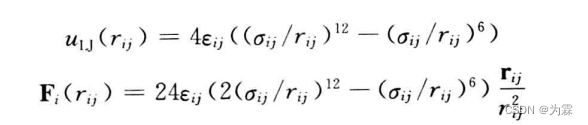
如果知道同种原子之间的Lennard-Jones势函数参数,可以利用混合规则估算两种不同原子之间的势函数参数,常用几何平均混合规则估算Aij和Bij

第二种形式的混合参数计算方法:
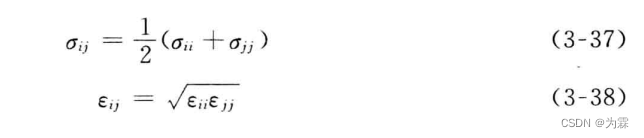
3.5.1 Lenard-Jones n-m势函数
- 在lenard-Jones势函数当中,指数12和6分别代表了排斥力的硬度和吸引力的范围。一般有机分子的r-6近似吸引力势函数,效果良好;但是r-12代表排斥力则势能太陡,因此除了调整A和B以外,还应当调整指数来调整引力的作用范围和排斥力的硬度。因此势函数的通式可以表达为:

- 实际应用中LJ12-10势函数近似为氢键,LJ 12-3势函数近似金属原子之间的作用
3.5.3 Morse势函数
Morse势函数具有三个势函数,其中Dc和r0分别对应LJ势函数的两个参数,b不与任何LJ参数对应,
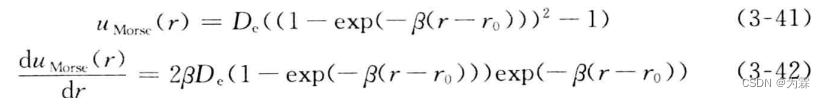
事实上,b参数是控制势函数的平坦程度,与LJ势函数中的n和m对应
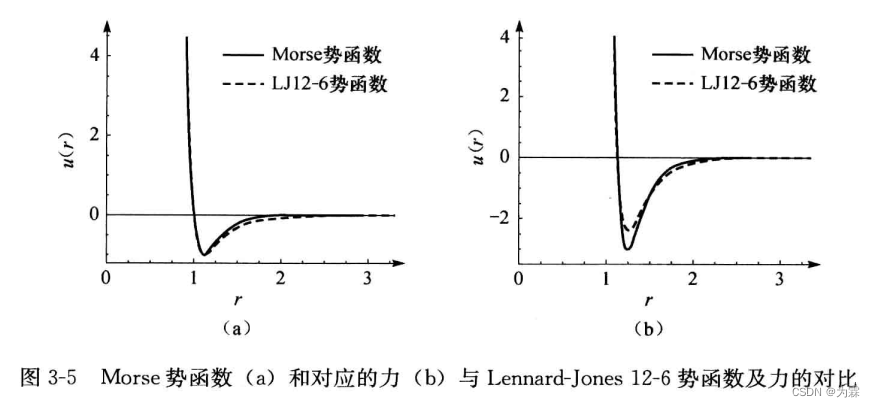
- morse函数和LJ函数几乎重合,最大差别在最大作用附近,morse函数更陡。对比如下


3.8 近程相互作用和远程相互作用
- 区别:近程相互作用距离相对较小,截去相距甚远的分子间作用力,不会产生太大误差,长程相互作用的截断近似则不成立
- 判断依据:势函数的渐进性质决定。势函数与rE-n具有相同的渐进性质的话,n越大,作用范围越小,n约小,作用范围越大。库仑力具有rE-1的渐进性质,典型的长程力;L-J相互作用具有r-6的渐进性质。n大于3为近程相互作用力,n小于等于3是长程相互作用。
- 长程和近程作用力本质上没有差别,但是MD对两种作用的计算方法完全不同
第四章 常用分子力场
分子力场——是一组描述分子体系之间相互作用的势函数,分为势函数的形式和参数集两个部分
势函数——只有少数几种形式,对应不同类型,形式是通用的
势函数参数——与具体的分子、原子有关,但不同分子或同一分子的不同部分,局部结构相同或相似,势参数常常可以通用
4.1 水分子力场
4.1.1 水分子实验性质
- 少数固体密度小于气体密度
- 液态水密度随温度下降增大,4摄氏度水密度最大 1g/cm3
- 水的高压固态相图异常丰富
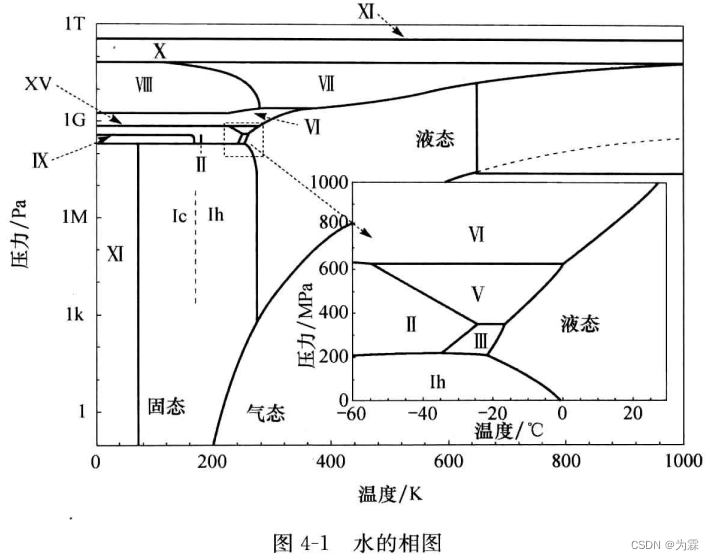
- 水的沸点和冰点


4.2 分子力场的分类
- 全原子力场:体系中的力点与原子意义对应
- 联合原子力场:将氢原子质量叠加到碳原子上形成联合原子,其他原子对氢原子的相互作用也叠加到碳原子上
- 粗粒度力场:把若干相关的原子当作一个整体力点的力场
- 反应性力场:由于大部分力场的成键相互作用用谐振子形式,当成键原子距离无限大时,相互作用势能也无限大,不允许化学键的断裂和形成;反应性分子力场中的相互作用势能除了与距离有关,还和键级有关。键级与成键原子距离和周围原子存在有关
4.3 MMn系列分子力场
- 适用范围:分子力场,可高精度对分子结构,生成含,振动频率
- 原子类型:数字序号表示;根据杂化类型,结构以及与之成键的原子类型有关
- 势函数形式:具体见书中p62页

4.4 全原子力场
生物和医药时MD最广泛应用领域。生物领域特点:
- 组成分子的结构单元简单,但顺序和链接方式一场复杂
- 必须在溶液当中
因此,生物分子力场必须有很好的可移植性和可扩展性
4.4.1 OPLS力场
- 势函数形式:键伸缩式和弯曲势均采用谐振子,范德华力采用LJ势函数12-6,静电势能采用库伦势
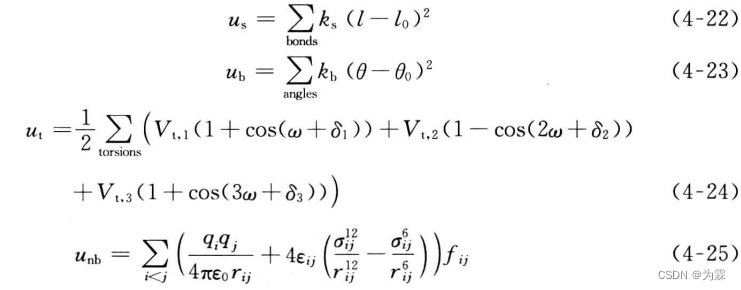
4.4.2 AMBER力场
- 势函数形势大部分与OPLS力场相同,但二面角扭曲势函数与范德华力势函数略有不同

- 原子类型:不单与杂化轨道类型有关,也和是否与电负性大的原子成键有关
4.4.3 CharMM力场
- 势函数形势:增加U-B相互作用,弥补键角弯曲势不足

4.5 联合原子分子力场
- 将CH4,CH3,CH2,CH,C当成一个联合原子或者力点,可以提高MD计算效率约一个量级
4.5.1 Gromos力场
- 成键相互作用

每一种形式都体现了对效率的精心设计;赝扭曲势是一个非常重要的势能项,保证了杂化碳原子的四面体结构和光学活性中心的错误消旋 - 范德华力相互作用

- 静电作用

- 原子类型:大多数氢原子被隐含在碳原子中,原子类型只有53中
4.5.3 CompASS力场
- 应用范围:材料科学领域的分子动力学模拟
- 势函数形式:

- 原子类型:原子元素符号,配位数,化学环境来分类
- 势参数:主要参照CFF力场
4.5.4 TraPPE和NERD力场
4.6 量子化学力场
4.6.1 MMFF94力场
- 用经验势函数拟合量子化学势能面得到的力场,而非拟合化合物结构和物性数据得到的力场
- 势函数形势:



- 原子类型:第一和第二个字符为元素符号,紧接着与该原子相连的原子名称、化学键种类
4.6.2 QMFF力场和CFF力场
QMFF是完全建立在第一性原理计算势能面基础上的分子力场,不包含实验参数;CFF是保留QMFF的经验势函数和参数,并用比例系数把QMFF力场转化为实际的CFF力场势函数
4.7 通用力场
大部分分子力场都具有很强的针对性,通用立场则适用于元素周期表中的大部分分子
,但是很难满足具有较高精读要求的模拟任务
4.7.1 Dreiding力场
- 原子类型:按照成键的杂化类型或几何构型进行分类
- 势函数形势:

4.7.2 UFF力场
- 原子类型:以原子的杂化类型、几何构型、氧化态和所处的化学环境等因素决定原子类型
- 势能构成

第五章 分子体系的运动方程及其数值解
5.1 分子体系的运动方程(牛顿第二定律)
分子体系的运动方程可以写成:

5.2 分子体系的运动方程(哈密顿运动方程)
- 哈密顿函数 H=K+U,其中K为体系总动能,U为总势能,仅与坐标位置有关,不包含时间
- 总动能计算公式:
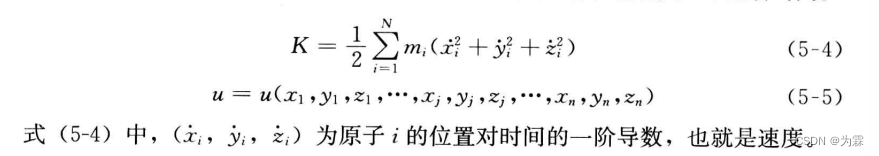
- 将哈密顿函数写成动量表达的形式为:
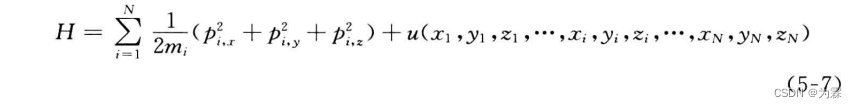
- 对于分子体系,将组成分子的各个原子的笛卡尔坐标变成变量计算并不方便,更方便的是用分子的质心坐标确定分子的质心位置,欧拉角确定分子取向,分子内坐标确定分子内原子的相对位置。因此实际计算过程中,通常不区分质心坐标、欧拉角和分子内坐标,统称为广义坐标,用qi表示。f是广义坐标的数量,也就是自由度。不受约束的N个自由质点的自由度为 f=3N。有r个完整约束时,自由度为3N-r
- 定义f个广义坐标矢量组成的q矢量,和f个广义坐标动量矢量组成的p矢量,体系总势能可以写成:

- 哈密顿方程是广义坐标和广义动量为独立变量的运动方程,由2f个一阶微分方程组成,与牛顿方程相比,优点有:
- 牛姐3N个二阶微分方程比求解2f个一阶微分方程困难
- 哈密度可以处理不受约束和约束的体系,而牛顿方程只适合处理不受约束的力学体系
- 哈密度方程是理解恒温恒压MD模拟的基础
5.3 常微分方程的数值解
5.3.1 差分方程
- 根据一阶常微分方程的初值问题:
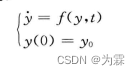
- 将时间变量t离散化,取步长为h,令
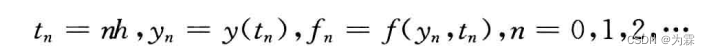
- 利用不同的差商近似f(y,t),并以fn的线性组合近似f(yn,tn),可以得到不同的差分公式:
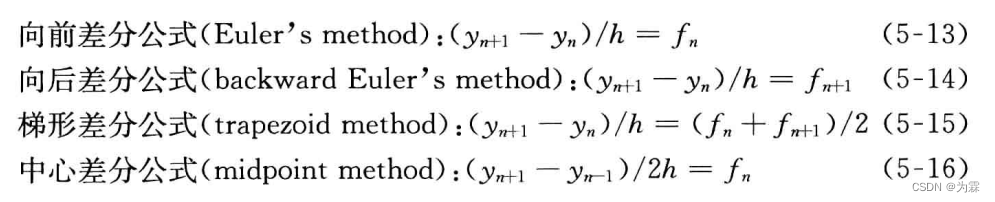
5.3.2 截断误差
将yn+1按照taylor公式展开:
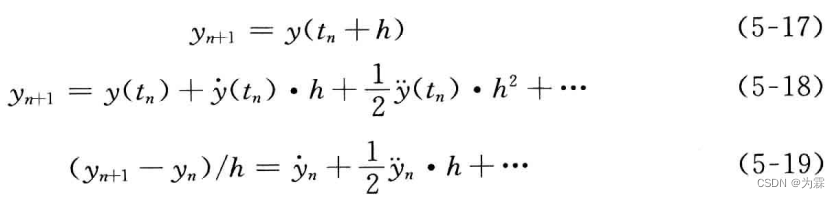
由此可以得到向前差分公式在tn点的截断误差:
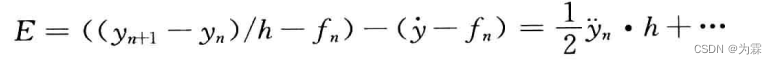
当h→0时,误差按h的一次方趋向于0,E=O(h)。同样的方法可以得到后差分公式的截断误差也按h的一次方趋向于0。但梯形差分公式以更快的速度趋向于0,E=O(h2)
5.3.3 利用差分公式解微分方程
将差分公式换算成以下形式,用于求解常微分方程的初值问题:
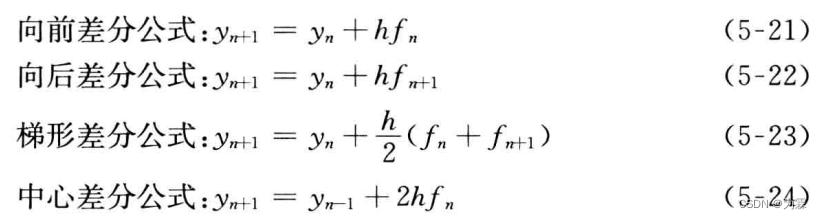
利用前差分公式,只需要知道处置y0,再计算f0=f(y0,t0),就可以计算y1和f1,依次计算y2,y3,,,最后得到任意时间的yn。向后差分公式,为了计算yn+1必须先计算fn+1,但计算fn+1必须先计算yn+1,称之为隐式差分公式。
5.4 分子体系运动方程的求解思路
(1)建立哈密顿方程组:
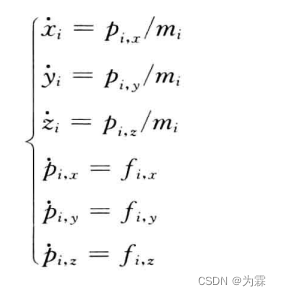
(2)确定运动方程组的初始条件,即初始时刻的原子坐标和速度,对于任何分子体系,只要确定了体系的初始条件和运动方程,体系随时间的演化就被确定
(3)选择适当的差分公式,计算下一刻的坐标和速度
(4)重复上述过程
5.5 分子体系的数值解
5.5.1 Euler算法
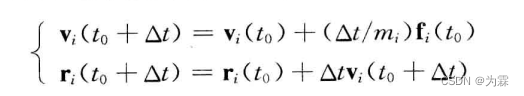
- 特点:单步、显式、一阶精度
5.5.2 Verlet算法
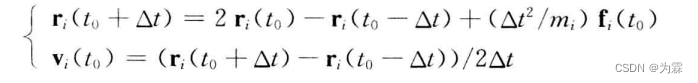
- 特点:两步差分法;坐标位置精读为四阶,速度精度为二阶;缺点,坐标位置由两个大数差值和一个小数求和得到,会导致计算精度降低
5.5.3 蛙跳算法
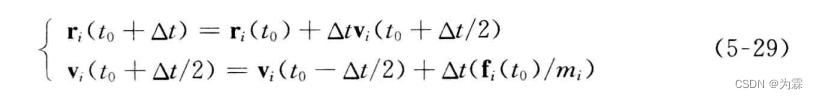
- 特点:速度跳过了t0时刻的计算,位置也跳过了to+h/2时刻的算法,计算速度和位置的精度都是四阶
5.5.4 速度Verlet算法

5.6 刚体运动方程的解
第六章 分子动力学模拟的技巧
6.1 周期性边界条件
6.1.1 周期性边界条件的物理内涵
- 由于模拟计算能力的限制,即使体系有10E10个分子,元胞的变成仍然只有0.67um左右,具有边界效应
- 消除边界效应,在MD模拟时引入周期性边界条件,模拟体系成为无限体系的一部分,称之为中心元胞
- 如图6-1所示,中心元胞中的任何一个粒子ri=(xi,yi),其像粒子周期性出现,位置为:
ri’=(xi+lL,yi+lL)
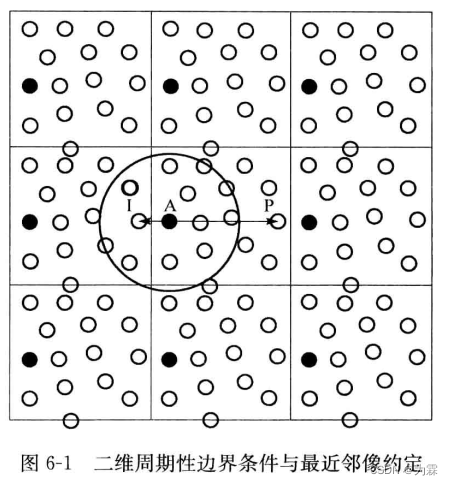
- 周期性边界条件的作用:当一个粒子离开中心元胞的时候,会有一个像粒子进入中心元胞,保证中心元胞的种类和数量在模拟过程中不变,进而消除边界效应
6.1.2 周期性边界条件的实现
- Fortran编程语言的实现:

- 计算周期性边界条件的时候,除了计算中心元胞中其他粒子对该粒子的作用,还需要计算像元胞中其他像粒子和粒子之间的作用,因此必须引入最近邻像约定
6.1.3 周期性边界条件的缺点
- 会给模拟体系强加一个周期性,引起长程有序,因此,中心元胞应当有足够的长度
- 存在长程力的话会给各向同性的模拟体系强加与中心元胞相同的对称性
- 抑制了长波的涨落,不能模拟出于气-液平衡、临界点等附近的性质
- 模拟合成高分子、生物大分子,必须保证中心元胞的尺度大于这些大分子的尺度。也要保证所模拟的大分子和其他像分子之间有足够的溶剂分子,否则将导致模拟的失败
- 为了验证MD结构可靠性,应当保持模拟体系密度不变的条件下改变模拟体系所包含的分子数目,进行多次模拟
6.2 势函数的计算技巧
两体、三体、四体相互作用分别与Ne2,Ne3,Ne4成正比。因此,提高MD模拟效率的核心在于提高计算力点之间的相互作用效率
6.2.1 多体相互作用的有效两体势近似
系统的总势能可以写成:
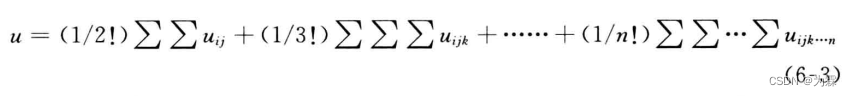
- 两体势只与两个分子之间的距离有关,与其他原子分子无关;三体势能除两个分子之间的距离以外,还与别的原子位置有关,需要两重以上的循环才能得到体系总势能,计算量大大增加
- 因此,出于计算效率及三体以上的相互作用相较于两体相互作用更为微软,势能可以近似为:

- 成为有效两体势。物理意义是i和j粒子处在其他粒子占有平均位置时的有效相互作用,表示对复杂分子间作用力的一种近似
6.2.2 最近邻像约定
- 定义:任意一个粒子,只与最邻近的粒子或者像粒子发生相互作用
- 如何实现:
- 将粒子在每个坐标方向施加邻像变化,得到两个粒子之间的最近像距离:(1)小于中心元胞对角线的一半(2)只计算距离最近的那一对像粒子
6.2.3 势函数的截断近似
- 近程相互作用,仅计算与粒子距离小于截断半径rc且位于截断球内的粒子贡献;注:rc不能超过中心元胞最小边长的1/2
- 用平均密度近似法对相互作用势能进行长程修正。注:该方法不能修正库伦势能
6.2.4 近邻列表算法
- 考虑粒子每一步只移动很小的距离,Verlet引入近邻列表,把r1>rc的粒子全部罗列到近邻列表当中,紧接着的若干步内置计算粒子与列表内粒子的距离。可以减少粒子距离计算
- 若干步后更新列表
6.2.5 格子索引法
- 将元胞划分为mXmXm个小格子,模拟只要计算粒子所在格子和相邻26个格子的粒子相互作用力。注意,格子的边长需大于截断半径rc,且m>3
- 计算时,先确定所有粒子的位置确定其归属的格子,并进行相互作用力计算
6.2.6 多步算法
- 近邻粒子被分为两类:(1)rp范围内,贡献较大的粒子(2)rp范围外,贡献较小且变化随时间变化较慢的粒子

6.3 长程力计算
6.3.1 周期性边界条件下的库伦势
周期性边界条件下Ewald计算库伦势方法:

6.3.2 Ewald算法原理
- 第一组点电荷,在每个电荷周围引入一个虚拟的电荷分布,用于屏蔽原来的电荷:

- 第二组虚拟电荷分布与第一组虚拟屏蔽电荷的绝对值完全相同,正负性相反
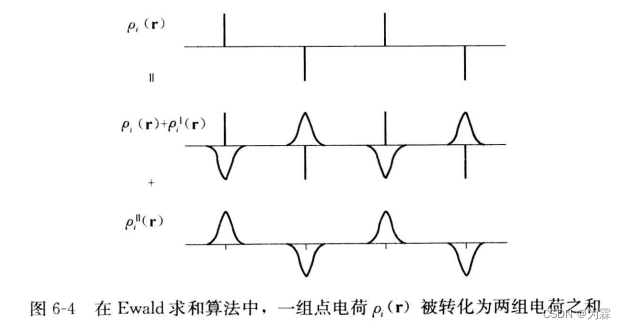
- 这样的处理不引入任何近似,但反而会加快库伦势的收敛
6.3.3 反应场算法:
- 作用在一个偶极矩i上的力被分为近程相互作用和长程相互作用。在截断半径外的偶极矩被近似为介电常数固定的连续介质,并在反应球内的形成一个反应场。则作用在偶极矩i上的反应场强度计算公式如下:

6.4 温度和压力控制技术
6.4.1 恒温MD模拟
Berendsen热浴法:
-
MD体系温度由体系总动能决定:
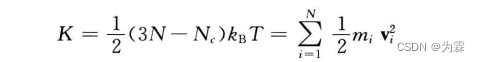
-
使模拟系统以
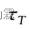 时间接近热浴温度,则:
时间接近热浴温度,则:
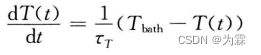
-
可以求出热浴法的速度调整系数为:
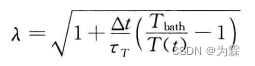
Nose-Hoover的扩展系数法
- 每个粒子运动方程引入一个外加耦合项,来调控体系温度:
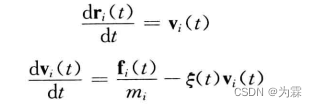

6.4.2 恒压MD模拟
- 比例系数恒压

- Andersen扩展体系

第7章 MD模拟的统计力学基础
第8章 第一性原理分子动力学模拟
第9章 分子动力学模拟的应用
9.1 MD模拟的计算机软硬件
9.2 模拟体系分子模型的建议
9.2.1 物理化学过程的参数空间
- 温度、压力等状态变量
- 电磁场和重力场等物理场变量
- 时间参数:由于MD模拟方法等模拟的现实世界物理化学过程的时间跨度为1ns~1ms,因此应当采取适当措施加速物理化学过程的进行
MD模拟的限制 - 当分子间平均距离大于分子间相互作用范围时(尤其大于截断半径时),MD模拟过程会忽略气体分子间的相互作用力,因此不适合稀薄的气体或者液体等稀少事件的物理化学过程
- 当模拟固体性质或者固体中发生的物理化学过程时,固体中的原子在模拟时间内很少发生长距离的迁移,导致模拟结果的统计偏差很大
- 不允许化学键的断裂和分解,难以研究高温条件下发生的物理化学过程
9.2.2 化学模型的建立
- 当中心元胞包含生物大分子或者合成高分子时,任意方向这些分子的尺寸都不能超过中心元胞的尺度;分子与像分子之间的距离必须大于分子间力的截断距离,保证在MD模拟过程中一个分子与其像分子之间的化学和物理分离,不存在任何相互作用
- 应当适当确定合成高分子的聚合度,不能太低,保证模拟结果可靠性,不能太高,影响模型顺利进行
- 模拟体系的总分子数量由中心元胞的空间尺度和模拟体系的总规模决定。首先,从物理层面讲,模拟体系应当大于实际体系的特征尺度(如超分子结果或者有序结构);技术方面,应当考虑计算资源的限制
9.2.3 分子力场模型的建立
- 全原子力场适用范围:无机分子、有机分子、生物分子、溶液、熔融盐
- 联合原子力场:有机分子、生物分子、合成高分子
- 粗粒度力场:脂质体、表面活性剂溶液、液晶
- 可极化力场:水溶液体系电离现象
- 势参数:
- 选用已被广泛采用的分子力场
- 如果缺少势参数,如何定制?
- 用量子化学、经验方法和红外光谱数据确定
- 当选用不同来源的力场参数集时:不同来源力场参数,在划分不同的运动模式时采取的方案不同,各参数不能自洽,需要格外小心
- 确定参数后,要优化和检查。成键作用参数决定分子的形状和形貌,如果偏离平衡状态要适当检查;非成键作用决定密度、沸点,应当适当调整
9.3 MD模拟的初始条件
要选择接近平衡状态的初始构型和初始速度,否则会影响模拟过程的稳定性:
(1)单原子分子和小分子体系,只要温度不太低,一般容易达到平衡状态,因此可以随机设定体系的初始构型。但是应当在MD模拟前优化体系的构型,降低体系势能,保证分子体系不是高度扭曲的构型,进而确保模拟过程的正常进行
(2)对于晶体和玻璃态物质,必须确保初始构型处在平衡附近
(3)对于合成高分子、生物大分子要根据大分子和溶剂的特征来确定初始构型
(4)初始速度通常可以根据Maxwell速度分布随机设定体系中各原子的初始速度
9.4 MD技术参数的确定:
- 数值积分算法或者差分格式,例如:Verlet蛙跳算法、速度蛙跳算法
- 数值积分时间步长
- 计算分子间相互作用力的算法,如计算库仑力的Ewald求和算法,计算非键相互作用的截断半径及其截断处理方法等
- 计算分子间相互作用力的节省时间算法,如近邻列表算法、 格子算法及相关参数等
- 模拟系综及其算法,模拟的统计系综(NVT/NPT等),状态变量P,V,T,实现统计系综的算法
- 模拟过程参数:包括准备和产出阶段的模拟步数、出错处理方法
- 模拟输出开关:模拟过程中需要计算的各种物理量,算法及相关参数
9.5 MD模拟的过程
- 将各种参数写入输入条件
- 确定输出信息、输出频率、输出文件格式。体系的构型和速度信息,即相轨迹文件或历史文件
- 设置断点处理方案
- 从不同的初始条件反复多次重复模拟
9.6 MD模拟结果的处理
9.6.1 体系的结构信息
分子与其基准构型之间的均方偏差根(RMSD)
- 对计算对象构型和基准构型进行旋转对齐操作
- 确定对比原子集C,计算对象构型与基准构型的对比原子的均方偏差根
- 根据以下公式计算RMSD:

径向分布函数
- 物理含义:与A类中心原子相距为r,厚度为△r的球壳中发现B原子的概率,与B原子在整个模拟体系均匀分布时的概率之比
- 球壳的体积计算公式:

- 物理意义:分子体系结构的一个重要函数,也是一个可以通过X射线衍射或者中子衍射实验方法 测量的函数
9.6.2 热力学性质
- 体系总动能:
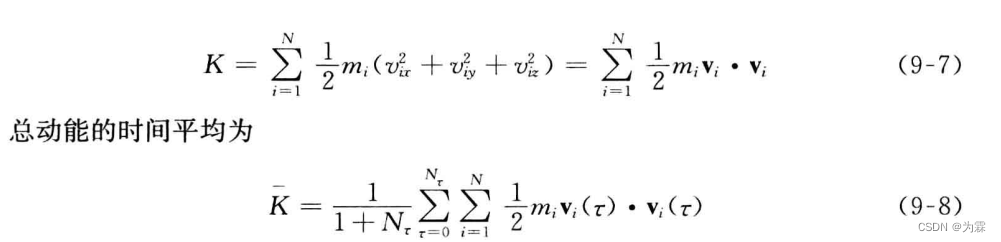
- 体系总势能:

- 总热力学能事总势能和总动能之和
- 根据总动能可以计算出体系的温度

- 压力通过维力定理计算得到:

- 由于
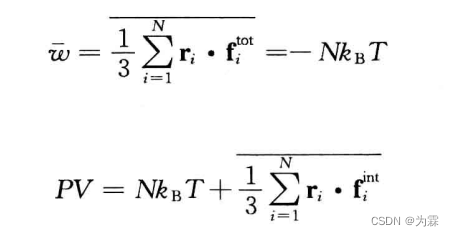
9.6.4 热力学涨落与热力学参数

- 正则系综的能量涨落与恒容热容
- 正则系综的压力涨落与等温压缩系数
- 正则系综的压力和势能的涨落与热压力系数
- 微正则系综中的涨落与热力学量的关系
- NPT系综中的涨落与热力学量的关系
9.6.5 迁移性质的计算
- 时间相关函数
- 速度自相关函数:模拟体系中特定的原子、分子的当前速度与早先某一时间的速度的相关程度

- 均方位移















 888
888











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









